NiO 支撑In2 O3(110)表面CO2加氢合成甲醇的理论计算研究
2021-12-14张科文陈毅飞胡廷平吕喜梅
张科文,陈毅飞,胡廷平,*,吕喜梅
(1.武汉轻工大学 化学与环境工程学院,湖北 武汉 430023;2.天津大学,石油化工技术开发中心,绿色合成与转化教育部重点实验室,天津 300072)
将二氧化碳加氢转化为高附加值的化学品或液体燃料是二氧化碳资源化利用的有效方法,不仅可以实现大气中的碳循环,缓和全球气候变化问题,还能够减少人类对化石能源如煤、石油和天然气的依赖,因此,受到世界范围内的广泛关注[1]。通过催化加氢将CO2转化为甲醇被认为是CO2转化最可能的利用途径,对高活性、高选择性催化剂的研究开发是实现CO2加氢合成甲醇的研究重点,已成为该领域富有挑战性的热点课题。其中,利用金属和金属氧化物作为催化剂加氢转化CO2合成甲醇得到了广泛的研究[2-9]。
CO2加氢合成甲醇催化剂主要有铜基催化剂和贵金属催化剂[1,9]两大类,Cu 基催化剂上合成甲醇的副产物多,甲醇产率低[10]。与Cu-ZnO-Al2O3催化剂相比,In2O3催化剂因其甲醇选择性高、几乎无CO 杂质生成而引起了研究人员的关注[11-13]。有研究表明,In2O3表面的氧空位在CO2加氢转化过程中对CO2的吸附和活化起着关键作用[14-17]。Otsuka 等[18]首先报道了在In2O3催化剂上CO2制备CO 的研究,随后Bielz 等[19]报道了In2O3催化剂在300 和340 K 下能被CO 和H2还原成金属铟。Sun 等[20]研究表明,CO2吸附在In2O3表面上的活性要比在Ga2O3表面上的活性高,并证实In2O3是一种很有前途的CO2转化催化剂。后期他们的实验结果表明,温度和压力对CO2加氢合成甲醇产率影响显著,产物主要有甲醇和CO,其产率高于许多其他文献报道的催化剂[21]。Oliver 等[11]研究发现,基于In2O3的催化剂在工业相关的CO2加氢反应条件下具有100%的甲醇选择性和出色的稳定性,这与工业的Cu-ZnO-Al2O3催化剂生成了鲜明的对比。Ye 等[15,16,22]根据DFT 计算指出,完美的In2O3(110)表面没有CO2加氢活性,而在缺陷In2O3(110)表面是CO2活化和甲醇合成的活性表面,且甲醇产率与In2O3(110)表面氧空位浓度呈现出正相关性。他们证明了In2O3表面的氧空位是活性位点,而甲醇是由CO2通过HCOO 路径生成的。在HCOO 路径中,吸附的CO2物种首先氢化为HCOO,然后与表面H 原子反应生成H2COO,甲氧基(H3CO)是通过H2COO 和H 原子的反应生成的,最后通过H3CO 的质子化反应合成甲醇[15,16]。
Ni 物种被证明可以与ZrO2载体产生相互作用促进氧空位的产生,从而对C=O 加氢活性具有促进作用[23-25],被镍改性之后的催化剂能增强对H2分子的吸附能力[26]。在Ni 掺杂的Fe 催化剂上,研究者也发现添加少量金属Ni 能够促进CO2加氢转化乙烯[27]。Jia 等[28]采用湿法化学还原法制备了In2O3负载镍催化剂,用于二氧化碳选择性加氢制甲醇,实验发现In2O3催化剂上Ni 活性位与表面氧空位之间的有效协同作用使CO2加氢具有较高的甲醇选择性。然而,Ni 对In2O3催化剂氧空位的产生有何影响,以及NiO 支撑的In2O3表面上CO2加氢合成甲醇的反应机理尚未见报道。因此,本研究拟对在NiO 支撑的In2O3(110)缺陷表面上CO2加氢合成甲醇过程进行研究,探究NiO 支撑对反应过程的影响。
1 计算部分
所有计算均是在Materials Studio 软件的DMol3模块中进行的[29]。表面吸附构型优化采用广义梯度近似(GGA),非局域相关能采用Perdew-Burke-Ernzerh(PBE)交换关联势进行描述,将基组设定为双数值加极化函数(DNP)。针对具有磁性的NiO,通过考察自旋多重度(multiplicity)对体系的影响,发现其影响非常小,同时考虑到计算量,因此,本文将不予考虑。对所有的原子采用自旋无限制计算的密度泛函半核伪电势(DSPP)方法,布里渊区格点k 点由Monkhorst-Pack method 方法生成,设置为2×2×1[30,31]。几何优化和能量计算的收敛准则为自洽场(SCF)1.0×10−6Ha、能量1.0×10−5Ha、最大力0.002 Ha/Å和最大位移0.005Å。
本文研究选择最稳定的In2O3体心立方晶系,并以相对稳定的In2O3(110)表面作为研究表面[21]。完美的In2O3(110)表面采用超晶胞建模[32],优化后的NiO 支撑的In2O3超晶胞的尺寸为10.29 ×14.55×21.04 Å。在图1(a)中,NiO 支撑的In2O3(110)完美表面(P 表面)由24 个In 原子,八个Ni 原子和48 个O 原子组成,分布在四层原子中,底层的八个In 原子替换成Ni 原子[33,34]。为防止因周期性边界条件而产生的晶面间相互作用,晶面上方真空层厚度设定为15 Å。如图1(b)所示,在NiO 支撑的In2O3(110)完美表面去除一个表面氧原子O3,即产生NiO 支撑的In2O3(110)缺陷表面(D 表面)[30,35]。在所有的计算中,最下面的两层原子被固定在晶胞的平衡位置,上面两层原子和吸附物种可自由弛豫。
吸附物种的吸附能Eads定义为:

式中,m 代表吸附物种,s 代表NiO 支撑的In2O3(110)表面。Em/s、Em和Es分别表示被吸附物种吸附于表面时的总能量、自由分子(自由基)的能量和D 或P 表面的总能量。
使用完全线性同步转移/二次同步转移(LST/QST)方法,在与反应物和产物相同的理论水平上进行沿着反应路径的过渡态搜索,最终的过渡态通过频率分析确认[36],保证过渡态有且只有一个虚频。反应能Er计算如下:

活化能垒Ea定义如下:

式中,EIS、ETS和EFS分别是指初始状态、过渡状态和最终状态的总能量。
2 结果与讨论
2.1 CO2加氢合成甲醇:HCOO 路径
如图1(b)所示,NiO 支撑的In2O3平板结构由三层In2O3层和一层Ni 层组成,表面包含两条具有四个In 原子和六个O 原子的In−O 链,NiO 支撑的In2O3(110)D 表面的氧空位是通过去除表面氧原子而产生的。研究表明,通过H2或CO 的还原,在In2O3催化剂表面上容易产生氧空位[11,19],因此,在实际反应条件下In2O3已经存在氧空位。图2显示了在NiO 支撑的In2O3(110)D 表面上由CO2加氢合成甲醇所涉及的中间体的优化结构,吸附能和几何参数列于表1。H2在In2O3(110)表面上解离成H 原子[1],CO2分子以多齿构型的形式填补氧空位,吸附能为−0.85 eV,其中,In2−Oa、In2−Ob和In3−C 的键长分别为2.25、2.31 和2.20 Å。在下文的叙述中,笔者以“氢化”指H 原子进攻吸附物的C 原子生成C−H 键,并以“质子化”指H 原子进攻吸附物的O 原子生成O−H 键。
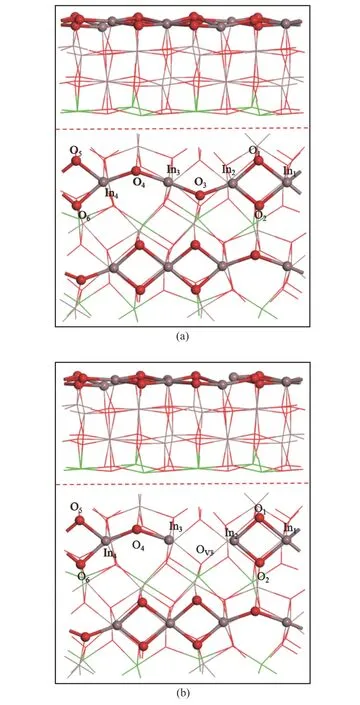
图1 (a) NiO 支撑的In2 O3(110)完美表面,(b) NiO 支撑的In2 O3(110)缺陷表面;侧视图(上),俯视图(下);其中,红色,O 原子;棕色,In 原子;绿色,Ni 原子Figure 1 (a) NiO supported In2 O3(110) perfect surface;(b) NiO supported In2 O3(110) defect surface;side view (upper),top view(lower);Here,the red O atom;Brown,In atom;Green,Ni atom
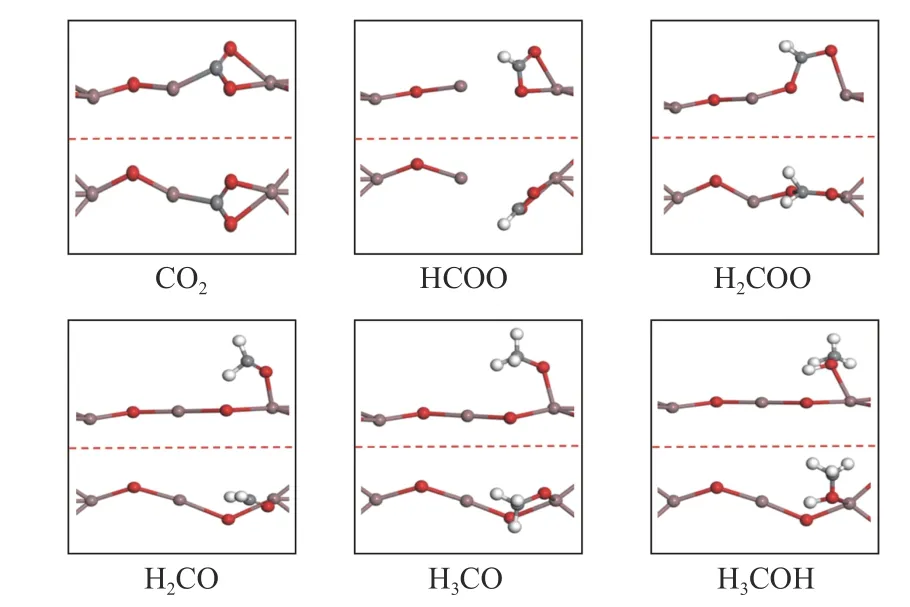
图2 在D 表面上CO2加氢合成甲醇中间体的优化结构,侧视图(上),俯视图(下)Figure 2 Optimized structures of CO2 hydrogenation to methanol intermediates on D surface,side view (upper),top view (lower)
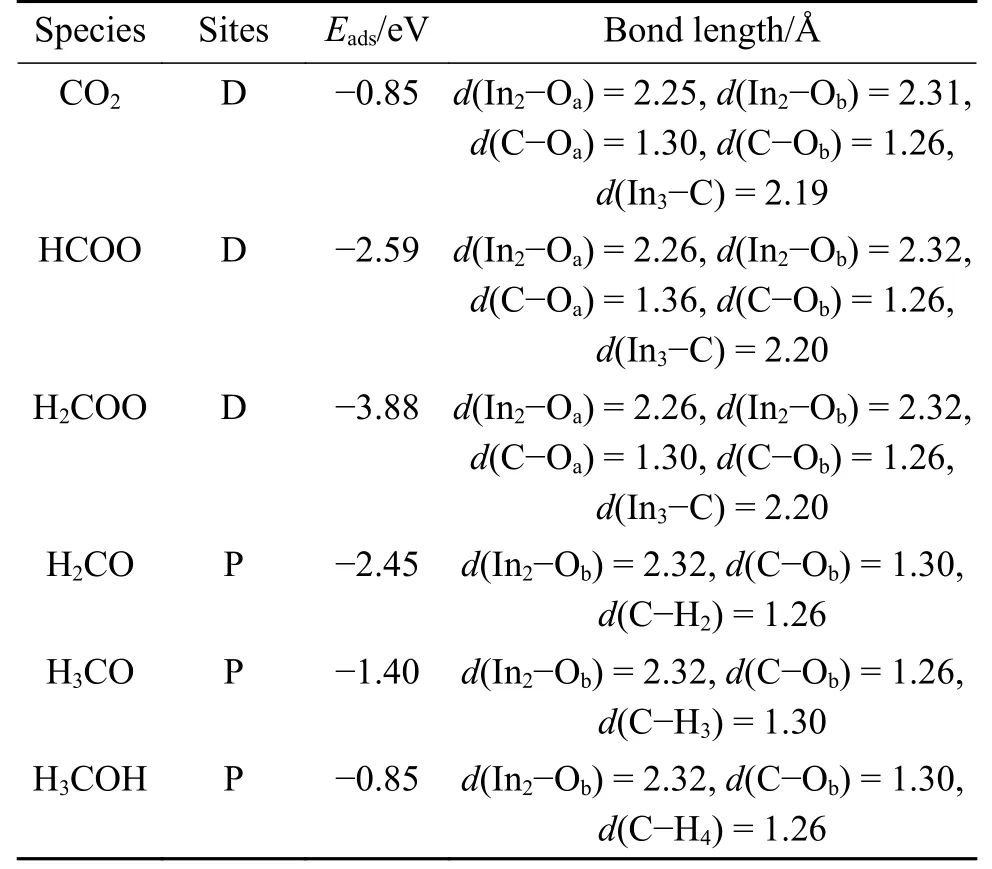
表1 在D 表面CO2加氢合成甲醇中间体的吸附能Eads和几何参数Table 1 Adsorption energies and geometric parameters of the intermediates of CO2 hydrogenation to synthesize methanol on the D surface
在D 表面,计算了CO2通过HCOO 路线加氢合成甲醇的过程。CO2分子首先加氢生成HCOO,生成的HCOO 继续与H 原子反应生成H2COO、H3CO,最终H3CO 通过质子化反应得到甲醇。CO2加氢合成甲醇所涉及的基元反应如图3 所示,其相对活化能垒和反应能见表2。
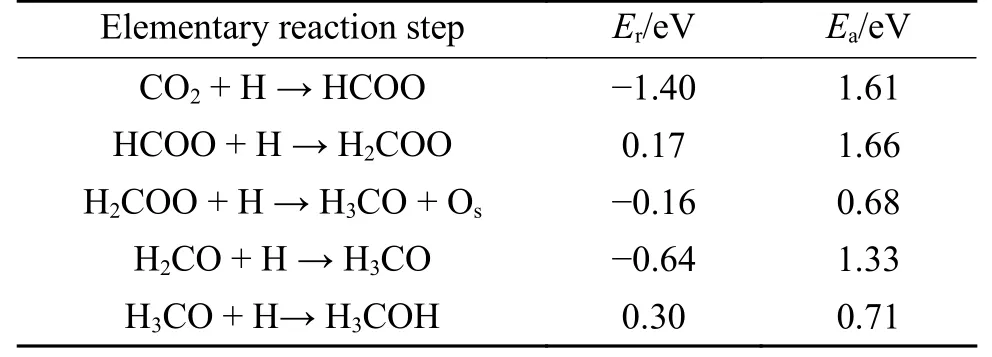
表2 D 表面上CO2加氢合成甲醇的各个基元反应的反应能Er和活化能垒EaTable 2 Reaction energy and activation energy barrier for the hydrogenation of CO2 to methanol on D surface
在CO2加氢合成甲醇的HCOO 路径中,吸附的CO2首先在D 表面氢化为HCOO,如图3(a)所示。在初始状态下,CO2与H 原子发生共吸附,C 和H 原子之间的距离为3.33 Å。在过渡状态下,C−H 距离缩短为2.19 Å,In3−C 键拉长并断裂。在最终状态下生成HCOO,In2−Oa、In2−Ob和C−H1的键长分别为2.26、2.32 和1.26 Å,该反应为放热反应(Er=−1.40 eV),活化能垒为1.61 eV。
在图3(b)所示的HCOO 加氢反应中,在初始状态下,HCOO 和H 原子共吸附在D 表面上,C 原子和H 原子之间的距离为2.63 Å,在过渡状态下,C−H 距离为1.02 Å,并生成In3−Oa键,在最终状态下,生成的H2COO 以多齿型吸附,该步反应为吸热反应(Er=+0.17 eV),活化能垒高达1.66 eV,表明在动力学上HCOO 在D 表面上的氢化被禁止,这意味着D 表面HCOO 加氢生成H2COO 可能是CO2加氢合成甲醇的速率控制步骤,此结果与Yang等[4,30]的计算结论一致。
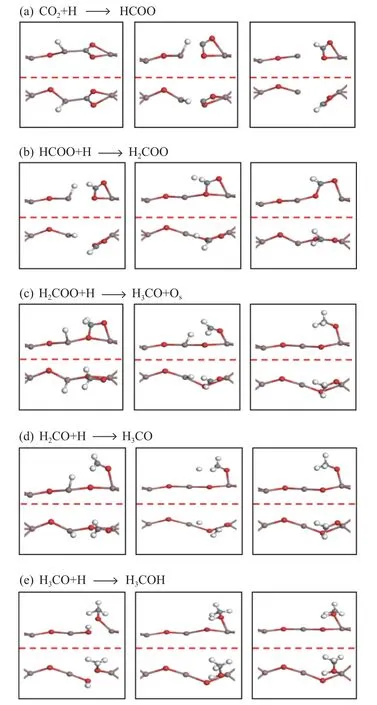
图3 D 表面CO2加氢合成甲醇的各个基元反应的初始、过渡和最终状态的优化结构;侧视图(上),俯视图(下)Figure 3 Optimized structure of the initial,transition and final states of the elementary reaction steps of the hydrogenation of CO2 to methanol on D surface;side view (upper),top view (lower)
H2COO 在D 表面的氢化存在两种路径。途径一,H2COO 直接氢化生成H3CO,如图3(c)所示。在初始状态下,H2COO 吸附在In3位连接的H 原子上,C 和H 原子之间的距离为2.80 Å。在过渡状态下,C−Ob键断裂,C−H 距离缩短为2.20 Å,而最终状态下产生的H3CO 以In2−O 键长为2.12 Å的形式吸附在In2位点,H2COO 直接加氢的活化能垒为0.68 eV,反应为放热反应(Er=−0.16 eV)。途径二,H3CO 是通过H2COO 转化成H2CO,H2CO 继续加氢生成的,如图3(d)所示。在初始状态下,H2CO 和H 共吸附在D 表面,C−H 距离为3.28 Å,在过渡状态下,C−H 距离缩短为2.12 Å,最终生成H3CO,该反应为放热反应(Er=−0.64 eV),活化能垒为1.33 eV。由此可见,H2CO 加氢比H2COO 的氢化需要翻越更高的能垒,H2COO 比H2CO 更容易生成H3CO,说明NiO 支撑的In2O3(110)缺陷表面能稳定H2COO,并防止其解离生成H2CO。因此,H3CO 主要是通过在NiO 支撑的有缺陷的In2O3(110)表面上由H2COO氢化而生成的。此结果与Ye 等[16]文献的计算结果有很大不同,Ye 等认为,H2CO 与H 生成HCOO 和H2的反应是自发进行的,H2CO 不能与表面H 原子一同存在。
H3CO 质子化生成甲醇的过程如图3(e)所示。在初始状态下,H3CO 与H 原子共吸附在表面,Ob−H 的距离为2.85 Å,过渡状态下Oa−H 和Ob−H的距离分别缩短为1.31 和1.19 Å,在最终状态下生成甲醇,并以−0.85 eV 的吸附能吸附在In2位点。该步反应为吸热反应(Er=+0.30 eV),活化能垒为0.71 eV,说明在动力学上,H3CO 通过质子化反应有利于形成甲醇。In2O3(110)缺陷表面随着甲醇的生成和脱附恢复成完美表面,这样使得反应可以在In2O3(110)表面上不断进行。
图4(a)所示为CO2在NiO 支撑的In2O3(110)D表面上由HCOO 路径合成甲醇的势能曲线,图4(b)所示为CO2在In2O3(110)D 表面上由HCOO 路径合成甲醇的势能曲线。CO2在In2O3(110)D 表面的反应步骤跟NiO 支撑的In2O3(110)D 表面的反应步骤相同。如图4(b)所示,从CO2加氢生成HCOO开始,在In2O3(110)D 表面上CO2和H 的共吸附能为−0.66 eV,该步反应的活化能垒为1.71 eV,过渡态为TS1。HCOO 继续加氢生成H2COO,该步反应为吸热反应(Er=+0.09 eV),活化能垒为1.73 eV,过渡态为TS2。HCOO 加氢生成的H2COO 可以与表面H 原子加氢生成H3CO,其活化能垒为0.74 eV,过渡态为TS3;H2COO 也可以转化成H2CO,进一步氢化成H3CO,需要克服1.35 eV 的能垒,其过渡态为TS4。最后,甲醇是由H3CO 质子化反应生成的,该步反应为吸热反应(Er=+0.28 eV),其活化能垒为0.64 eV,过渡态为TS5。
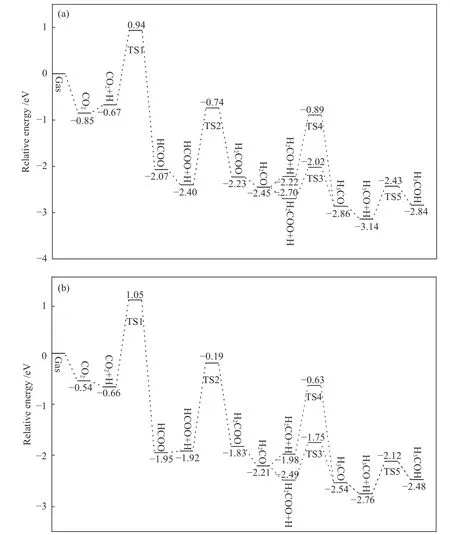
图4 D 面上HCOO 路径合成甲醇的势能曲线,(a) NiO 支撑的In2 O3(110) D 表面,(b) In2 O3(110) D 表面Figure 4 Potential energy for methanol synthesis through HCOO route on D surface
对比图4(a)和图4(b)可以看出,CO2在NiO支撑的In2O3(110)D 表面的吸附能比In2O3(110)D表面上的吸附能高,说明NiO 支撑的In2O3(110)D表面有利于CO2的稳定吸附和活化加氢生成HCOO,而HCOO 氢化生成H2COO 需要克服的活化能垒是整个反应步骤最高的,所以此步反应是CO2加氢合成甲醇在HCOO 路径中的速率控制步骤。
2.2 CO2加氢合成甲醇:RWGS 路径
CO2加氢反应生成CO 和H2O 称为逆水煤气变换(RWGS)反应,是有效利用二氧化碳并将其转化为化学资源的重要反应[37]。CO2解离生成CO,通过CO 加氢也是合成甲醇的一种路径,因此,本研究也研究了CO2在D 表面上的分解。图5显示了在D 表面由CO 加氢合成甲醇所涉及中间体的优化结构,其吸附能和几何参数列于表3。在D表面,CO 吸附在Ov3位上的吸附能为−0.77 eV,In2−C 和In3−C 的键长分别为2.44 和2.42 Å,甲醇最终通过CO 连续氢化合成的。D 表面上CO 加氢所涉及基元反应的初始态、过渡态和最终状态如图6所示,计算所得的活化能垒和反应能见表4。
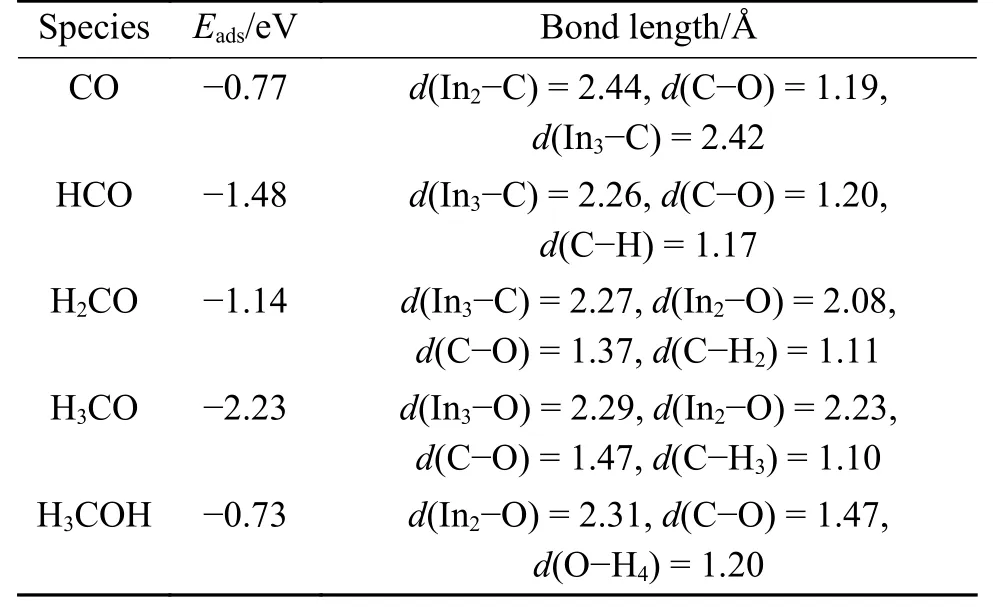
表3 D 表面CO 加氢合成甲醇中间体的吸附能Eads和几何参数Table 3 Adsorption energy and geometric parameters of methanol intermediates synthesized by hydrogenation of CO on D surface
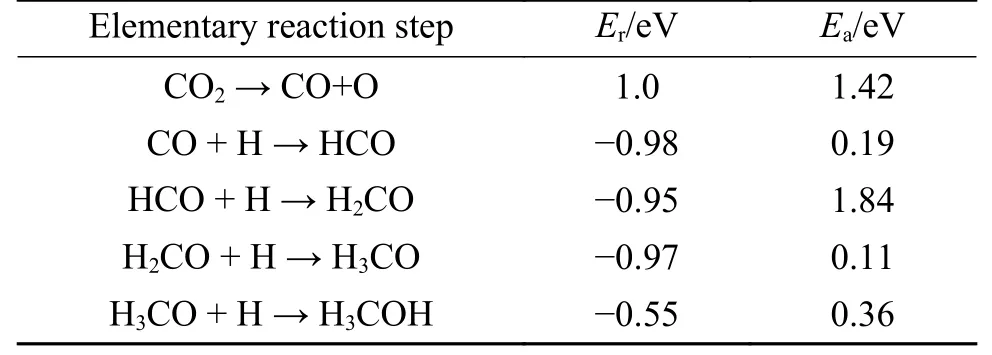
表4 D 表面CO 加氢合成甲醇的各个基元反应的反应能Er和活化能垒EbTable 4 Reaction energy and activation energy barrier for the hydrogenation of CO on D surface to methanol
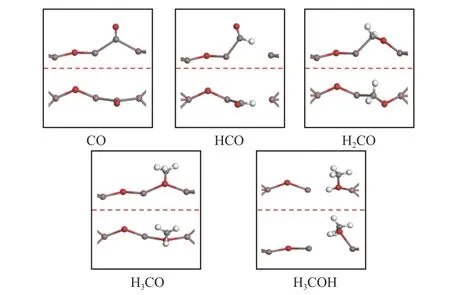
图5 在D 表面上CO 加氢合成甲醇中间体的优化结构,侧视图(上),俯视图(下)Figure 5 Optimized structure of CO hydrogenation to methanol intermediates on D surface,side view (upper),top view (lower)
D 表面上CO2直接解离为CO 的过程如图6(a)所示。在初始状态下,CO2吸附在Ov3氧空位上,在过渡状态下,In2−Ob键断裂,C−Oa键长为2.18 Å。在最终状态下生成的CO 吸附在In3位点,In3−C 键的键长为2.68 Å,该步反应是吸热反应(Er=+1.0 eV),活化能垒为1.42 eV。Dou 等[30]计算了CO2在ZrO2支撑的In2O3(110)缺陷表面上解离,活化能垒为2.16 eV,此步反应为吸热反应(Er=+1.35 eV)。因此,NiO 支撑的In2O3(110)缺陷表面相对于ZrO2支撑的In2O3(110)缺陷表面,CO2解离的活化能垒和反应能更低。
如图6(b)所示,在初始状态下,CO 与H 原子共吸附在D 表面,CO 吸附在In3位点的顶部,而H吸附在In2位点,C 和H 原子之间的距离为3.28 Å。在过渡状态下,C 和H 之间的距离缩短为2.32 Å,最终生成HCO,其In3−C 键长为2.26 Å,该步反应为放热反应(Er=−0.98 eV),活化能垒为0.19 eV,因此,在D 表面上CO 与表面H 原子的反应很容易。
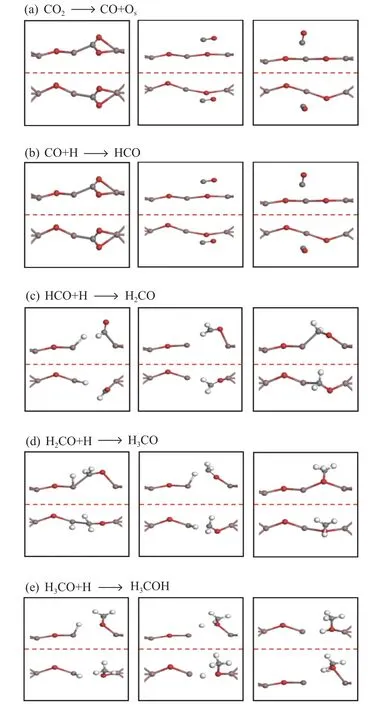
图6 D 表面CO 加氢合成甲醇的各个基元反应的初始、过渡和最终状态的优化结构;侧视图(上),俯视图(下)Figure 6 Optimized structure of the initial,transition and final states of the elementary reaction steps for the hydrogenation of surface CO to methanol;side view (upper),top view (lower)
如图6(c)所示为HCO 加氢生成H2CO 的过程。在初始状态下,HCO 和H 原子共吸附在D 表面,C 和H 原子之间的距离为2.38 Å。在过渡状态下,In2−C 键断裂同时形成C−H 键,C−H 键的键长为1.11 Å,最终生成H2CO,其In2−O 和In3−C 的键长分别为2.08 和2.27 Å,该步反应为放热反应(Er=−0.95 eV),活化能垒为1.84 eV。H2CO 与H 反应生成H3CO 的过程,如图6(d)所示。在初始状态下,H2CO 与H 共吸附在D 表面,C 和H 原子的距离为2.02 Å。在过渡态下,In3−C 键发生断裂生成H3CO,以H3CO 中的O 原子填充Ov3氧空位形成完美表面。最后是H3CO 质子化生成H3COH的过程如图6(e)所示。在初始状态下,H3CO 和H 共吸附在D 表面,O−H 距离为2.45 Å,最终状态下形成O−H键,其键长为1.20 Å,该反应是放热反应(Er=−0.55 eV),活化能垒为0.36 eV。
图7(a)所示为CO2在NiO 支撑的In2O3(110)D表面上由RWGS 路径合成甲醇的势能曲线,图7(b)所示为CO2在In2O3(110)D 表面上由RWGS 路径合成甲醇的势能曲线。如图7(b),CO2首先在In2O3(110)D 表面上解离成CO 和O,该反应是吸热反应(Er=+1.29 eV),其活化能垒为1.48 eV,过渡态为TS1。CO 与表面H 原子生成HCO 的反应为吸热反应(Er=+0.86 eV),其活化能垒为+0.24 eV,过渡态为TS2。HCO 继续加氢生成H2CO,该反应为放热反应(Er=−0.81 eV),活化能垒为1.89 eV,过渡态为TS3。H2CO 与表面H 原子进一步氢化成H3CO,需要克服的能垒为0.24 eV,其过渡态为TS4。最后,甲醇是由H3CO 质子化反应生成的,该反应为放热反应(Er=−0.47 eV),其活化能垒为0.43 eV,过渡态为TS5。
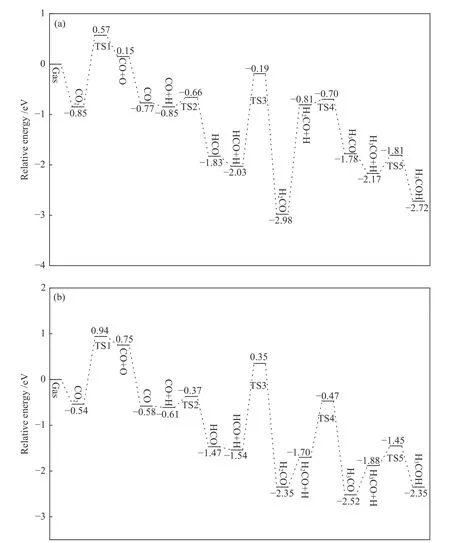
图7 在D 面上RWGS 路径合成甲醇的势能曲线,(a) NiO 支撑In2 O3(110) D 表面,(b) In2 O3(110) D 表面Figure 7 Potential energy for methanol synthesis by RWGS route on D surface
对比HCOO 和RWGS 路径发现,NiO 支撑的In2O3(110)D 表面上CO2解离成CO 的反应是比较困难的,而在RWGS 路径中CO2的解离反应不仅需要吸收大量的热量,还需要翻越很高的能垒。因此,CO2加氢合成甲醇主要是通过HCOO 路径,而不是RWGS 路径。此结果与Jia 等[28]的实验结果相同,Jia 等认为,CO2加氢反应里甲醇为主要产物,CO 为副产物,CO2倾向于加氢生成 HCOO路径,而不倾向于生成甲酸盐进而得到甲醇的路径。
2.3 电荷布局分析
Mulliken 电荷布局分析[35,38]是反映吸附体系中原子与键在吸附前后的电子分布。为分析二氧化碳在In2O3(110)D 表面和NiO 支撑In2O3(110)D表面上的吸附性质,对CO2分子在这两个表面的吸附构型进行Mulliken 电荷布局分析。如表5 所示,CO2吸附后,催化剂表面的In、Ni、O 原子及CO2中各原子电荷量发生变化,CO2电荷在催化剂作用下被重新分配。CO2在In2O3(110)D 表面和NiO支撑In2O3(110)D 表面的总电荷分别为−0.454 e 和−0.457 e,所有吸附在表面的CO2均得到电荷,表明CO2在氧化铟表面吸附时,CO2与氧化铟表面之间存在电荷转移,从而达到强化催化剂表面吸附和活化CO2。
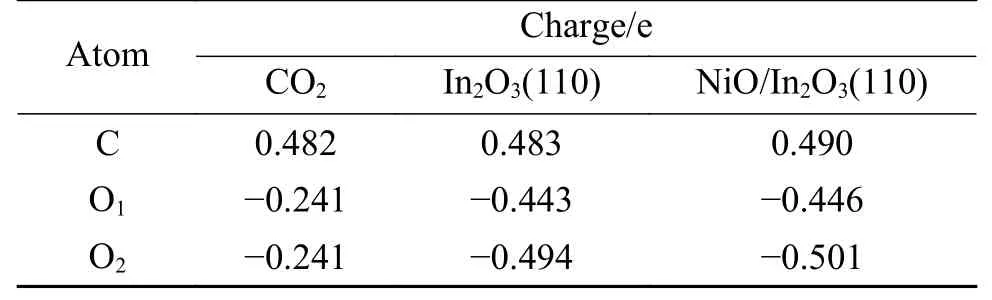
表5 CO2在In2 O3(110)D 表面,NiO 支撑In2 O3(110)D 表面吸附的Mulliken 电荷布局Table 5 Mulliken atomic charge populations for CO2 adsorption on the D surface,NiO supported In2 O3(110)D surface
3 结论
采用密度泛函理论(DFT),研究了在NiO 支撑的In2O3(110)缺陷表面上CO2加氢合成甲醇的反应机理。结果表明,NiO 支撑的In2O3(110)缺陷表面较In2O3(110)缺陷表面更稳定,NiO 支撑能够强化In2O3催化剂对CO2的吸附性能;Muliken 电荷布局分析也显示CO2得到电子,电荷从表面转移到CO2,从而强化吸附并活化CO2。CO2加氢合成甲醇主要是通过HCOO 路径进行的,D 表面上HCOO与表面H 原子生成H2COO 的活化能垒为1.66 eV,是整个HCOO 路径能垒最高的,为CO2加氢合成甲醇的速率控制步骤。NiO 支撑的In2O3(110)缺陷表面对CO2的加氢具有促进作用,有助于CO2沿着HCOO 路径合成甲醇,从而提高CO2加氢合成甲醇的效率,表明NiO 支撑的氧化铟是一种潜在的CO2转化利用催化剂。
