越婢加术汤的HPLC指纹图谱建立、含量测定及化学模式识别分析
2021-12-10赵峰高太祥石莉尧王瑞
赵峰 高太祥 石莉尧 王瑞

中圖分类号 R284.1 文献标志码 A 文章编号 1001-0408(2021)22-2724-07
DOI 10.6039/j.issn.1001-0408.2021.22.07
摘 要 目的:建立越婢加术汤的高效液相色谱(HPLC)指纹图谱,测定其中盐酸麻黄碱和盐酸伪麻黄碱的含量,同时进行化学模式识别分析。方法:以甘草酸铵为参照,使用《中药色谱指纹图谱相似度评价系统(2012版)》建立10批越婢加术汤的HPLC指纹图谱并进行相似度评价,结合混合对照品指认共有峰;采用HPLC法测定盐酸麻黄碱和盐酸伪麻黄碱的含量;采用SPSS 26.0软件进行聚类分析,采用SIMCA 13.0软件进行主成分分析和偏最小二乘法-判别分析,并筛选影响越婢加术汤质量的差异性成分。结果:越婢加术汤指纹图谱中共有20个共有峰,与对照指纹图谱的相似度均大于0.92;共指认了2个色谱峰,分别为甘草苷(峰11)和甘草酸铵(峰18)。盐酸麻黄碱、盐酸伪麻黄碱检测质量浓度的线性范围分别为0.98~48 μg/mL(r=0.999 9)、1.02~51 μg/mL(r=0.999 2);精密度、重复性、稳定性(24 h)试验的RSD均小于2%;平均加样回收率分别为105.67%(RSD=2.88%,n=9)、104.15%(RSD=2.02%,n=9);含量分别为0.008 1~0.014 3、0.002 5~0.011 8 mg/mL。聚类分析结果显示,10批越婢加术汤可聚为3类,S1聚为一类,S3聚为一类,S2、S4~S10聚为一类。主成分分析结果显示,S3位于得分图的最右侧,S1位于得分图的右侧,S2、S4~S10位于得分图的中部。偏最小二乘法-判别分析结果与聚类分析、主成分分析基本一致,且峰9、峰3、峰12、峰8、峰19、峰18(甘草酸铵)、峰13、峰20、峰11(甘草苷)的重要性投影值均大于1。结论:所建HPLC指纹图谱和含量测定方法操作简便、准确,结合化学模式识别可用于越婢加术汤的质量控制。峰9等9个成分为影响越婢加术汤质量的差异性成分。
关键词 越婢加术汤;高效液相色谱法;指纹图谱;含量测定;化学模式识别
Establishment of HPLC Fingerprints, Content Determination and Chemical Pattern Recognition Analysis of Yuebi Jiazhu Decoction
ZHAO Feng,GAO Taixiang,SHI Liyao,WANG Rui(School of Chinese Medicine and Food Engineering, Shanxi University of TCM, Shanxi Jinzhong 030619, China)
ABSTRACT OBJECTIVE: To establish the HPLC fingerprints of Yuebi jiazhu decoction, determine the contents of ephedrine hydrochloride and pseudoephedrine hydrochloride and carry out chemical pattern recognition analysis. METHODS: Using ammonium glycyrrhizinate as control, HPLC fingerprint of 10 batches of Yuebi jiazhu decoction was established and the similarity was evaluated with Similarity Evaluation System for TCM Chromatographic Fingerprint (2012 edition), and common peaks were identified in combination with mixed control. HPLC method was used to determine the contents of ephedrine hydrochloride and pseudoephedrine hydrochloride. SPSS 26.0 software was used for cluster analysis; SIMCA 13.0 software was used for principal component analysis and partial least squares discriminant analysis; the differential components affecting the quality were screened. RESULTS: There were 20 common peaks in the fingerprint of Yuebi jiazhu decoction, and the similarities with control fringerprint were all greater than 0.92. Two peaks were identified, which were liquiritin (peak 11) and ammonium glycyrrhizate (peak 18). The linear range of ephedrine hydrochloride and pseudoephedrine hydrochloride were 0.98-48 μg/mL (r=0.999 9) and 1.02-51 μg/mL (r=0.999 2), respectively. RSDs of precision, reproducibility and stability tests (24 h) were all less than 2%. The average recoveries were 105.67% (RSD=2.88%, n=9) and 104.15% (RSD=2.02%, n=9), respectively. The contents of ephedrine hydrochloride and pseudoephedrine hydrochloride were 0.008 1-0.014 3, 0.002 5-0.011 8 mg/mL. Results of cluster analysis showed that among the 10 batches of Yuebi jiazhu decoction, S1 was clustered into one class, S3 was clustered into one class, and S2, S4-S10 were clustered into one class. The results of principal component analysis showed that S3 was located at the far right side of the scoring plot, S1 at the right side of the scoring plot, and S2, S4-S10 at the middle of the scoring plot. The results of partial least squares discriminant analysis were basically consistent with the results of cluster analysis and principal component analysis. The variable importance projection values of peak 9, peak 3, peak 12, peak 8, peak 19, peak 18 (ammonium glycyrrhizinate), peak 13, peak 20 and peak 11 (liquiritin) were greater than 1. CONCLUSIONS: Established HPLC fingerprint and the method for content determination are simple and accurate. Combined with chemical pattern recognition, they can be used for the quality control of Yuebi jiazhu decoction. Nine components such as peak 9 are the differential components affecting the quality of Yuebi jiazhu decoction.
KEYWORDS Yuebi jiazhu decoction; HPLC; Fingerprint; Content determination; Chemical pattern recognition
越婢加术汤出自《金匮要略》[1],由麻黄、石膏、白术、生姜、甘草和大枣等6味药材组成,常用于风湿热痹、水肿、肾炎的臨床治疗[2-3]。此方中,麻黄解表发汗以消水肿,为君药;石膏性寒以清涤里热又牵制麻黄发越过猛,为臣药;白术健脾利湿并强麻黄利尿消肿之功,为佐药;再以生姜、大枣、甘草补中和胃并牵制麻黄发汗而耗伤津液[4];诸药合用,共奏发汗利水、清热除烦之功[5]。越婢加术汤主要含有生物碱类、黄酮类、挥发油类、三萜类、多糖类等有效成分,其中盐酸麻黄碱和盐酸伪麻黄碱为生物碱类成分,具有止咳平喘、发汗利尿的作用;甘草苷为黄酮类成分,具有抗抑郁、保护心脏和神经等活性;甘草酸铵为齐墩果烷型三萜类成分,具有抗炎、抗病毒、保肝、保护神经等活性[6-10]。由于越婢加术汤的化学成分复杂,加之药材产地、批次、煎煮方式的不一致,使得煎液质量不稳定,从而导致临床用药差异[11]。此外,目前关于越婢加术汤的研究多集中于其临床应用方面[12-13],且2020年版《中国药典》(一部)也未收载该方的质量标准,因此对越婢加术汤进行质量控制就显得尤为重要。
指纹图谱具有系统性、整体化的特点,能比较客观、完整地反映中药复方中尽可能多的化学成分信息,已被广泛用于中药复方的质量控制和评估领域[14-16]。化学模式识别是化学计量学的重要组成部分,包括聚类分析(CA)、主成分分析(PCA)、偏最小二乘法-判别分析(PLS-DA)等,其能通过变换和构造模型,使复杂数据简单化,最终实现对样品的分类[17]。基于此,本研究在参考2020年版《中国药典》(一部)[18]的基础上,建立了越婢加术汤的高效液相色谱(HPLC)指纹图谱,并测定了该方君药麻黄中盐酸麻黄碱和盐酸伪麻黄碱的含量,同时结合CA、PCA、PLS-DA进行化学模式识别分析,旨在为越婢加术汤的质量控制提供参考。
1 材料
1.1 主要仪器
本研究所用主要仪器有2695-2998型HPLC仪及配备的Empower 3色谱工作站(美国Waters公司)、AB135- S型电子天平(瑞士Mettler Toledo公司)、ZDHW型调温电热套(北京中兴伟业仪器有限公司)、HC-2518型高速离心机(科大创新股份有限公司中佳分公司)、KQ55200V型超声波清洗器(昆山市超声仪器有限公司)等。
1.2 主要药品与试剂
甘草酸铵对照品(批号53956-04-0,纯度94.20%)购自上海诗丹德标准技术服务有限公司;盐酸麻黄碱对照品(批号171241-201809,纯度100.0%)、盐酸伪麻黄碱对照品(批号171241-201809,纯度99.8%)、甘草苷对照品(批号111610-201607,纯度93.1%)均购自中国食品药品检定研究院;732型强酸苯乙烯阳离子交换树脂(粒径1 mm,批号C10716501)购自上海麦克林生化科技有限公司;甲醇(色谱纯)、乙腈(色谱纯)均购自北京百灵威科技有限公司,其余试剂均为分析纯,水为纯净水。
麻黄饮片、白术饮片、石膏饮片、甘草饮片、生姜和大枣经山西中医药大学中药学系裴香萍副教授鉴定,分别为麻黄科植物草麻黄Ephedra sinica Stapf的干燥草质茎、菊科植物白术Atractylodes macrocephala Koidz.的干燥根茎、豆科植物甘草Glycyrrhiza uralensis Fisch.的干燥根和根茎、姜科植物姜Zingiber officinale Rosc.的新鲜根茎、鼠李科植物枣Ziziphus jujuba Mill.的成熟果实。除生姜、大枣(生姜的产地均为山西,大枣的产地均为河北,均购自山西省晋中市各超市)外,其余饮片均购自山西省晋中市各药店及诊所。10批越婢加术汤中麻黄等4种饮片样品的来源信息见表1。
2 方法与结果
2.1 指纹图谱建立
2.1.1 色谱条件 以Hypersil BDS C18(250 mm×4.6 mm,5 μm)为色谱柱,以乙腈(A)-0.1%磷酸溶液(含0.04%三乙胺)(B)为流动相进行梯度洗脱(0~50 min,5%A→17.5%A;50~70 min,17.5%A→35%A;70~85 min,35%A→50%A);流速为1 mL/min;检测波长为230 nm;柱温为30 ℃;进样量为10 μL。
2.1.2 越婢加术汤的制备 由于原方中各饮片用量较大,因此根据各味饮片的常用剂量、配比功效以及前期药理研究获得的有效剂量范围进行调整,得到越婢加术汤的处方比例如下:麻黄9 g、石膏18 g、甘草6 g、大枣15 g、生姜9 g、白术9 g[19-21]。参照越婢加术汤古代煎煮方法,取水600 mL,加入麻黄煮30 min后,纳诸药大火煮沸再小火微沸煮至200 mL,即得越婢加术汤(每升药液含生药总量330 g)[1]。同法制得10批(编号为S1~S10)。煎煮时采用相同容器并结合重复性试验以保证制备过程的一致性。
2.1.3 供试品溶液的制备 取“2.1.2”项下10批越婢加术汤各4 mL,加甲醇6 mL,称定质量,超声(功率200 W,频率40 kHz)处理20 min,放冷至室温,再次称定质量,用甲醇补足减失的质量,摇匀,以14 000 r/min离心10 min,取上清液,经0.22 μm微孔滤膜滤过,取续滤液,即得供试品溶液。
2.1.4 混合对照品溶液的制备 精密称取甘草苷、甘草酸铵对照品,加甲醇溶解,制得甘草苷、甘草酸铵质量浓度分别为0.02、0.2 mg/mL的混合对照品溶液。
2.1.5 精密度试验 取“2.1.3”项下供试品溶液(编号S1)适量,按“2.1.1”项下色谱条件连续进样测定6次,以甘草酸铵为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,20个共有峰相对保留时间的RSD均小于1.5%(n=6),相对峰面积的RSD均小于2.5%(n=6),表明方法精密度良好。
2.1.6 重复性试验 取“2.1.2”项下越婢加术汤(编号S1),共6份,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,以甘草酸铵为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,20个共有峰相对保留时间的RSD均小于1.5%(n=6),相对峰面积的RSD均小于2.5%(n=6),表明方法重复性良好。
2.1.7 稳定性试验 取“2.1.3”项下供试品溶液(编号S1),分别于室温下放置0、2、4、8、12、24 h时按“2.1.1”项下色谱条件进样测定,以甘草酸铵为参照,计算各共有峰的相对保留时间和相对峰面积。结果显示,20个共有峰相对保留时间的RSD均小于1.5%(n=6),相对峰面积的RSD均小于2.5%(n=6),表明供试品于室温下放置24 h内稳定性良好。
2.1.8 指纹图谱的建立 取“2.1.2”项下10批越婢加术汤,按“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定,记录色谱图。将10批越婢加术汤的色谱图导入《中药色谱指纹图谱相似度评价系统(2012版)》进行分析,以S1为参照图谱(该批样品的色谱信息较多),采用中位数法,设置时间窗宽度为0.1 min,对色谱峰进行多点校正和全峰匹配,得到10批越婢加术汤的叠加指纹图谱和对照指纹图谱(R)。结果显示,10批越婢加术汤共有20个共有峰,详见图1、图2。
2.1.9 共有峰的指认 经与混合对照品色谱图[由于甘草苷、甘草酸铵与盐酸麻黄碱、盐酸伪麻黄碱的结构存在差异,无法采用相同的色谱条件进行检测,因此选择甘草苷、甘草酸铵为对照。取“2.1.4”项下混合对照品溶液,按“2.1.1”项下色谱条件进样,得到色谱图(图3)]比对,共指认了2个共有峰,分别为甘草苷(峰11)、甘草酸铵(峰18)。因甘草酸铵色谱峰峰面积适中、分离度较好,故以甘草酸铵为参照。
2.1.10 相似度评价 采用《中药色谱指纹图谱相似度评价系统(2012版)》对10批越婢加术汤样品进行相似度评价。结果显示,与对照指纹图谱(R)相比,10批样品的相似度均大于0.92,表明10批样品的质量差异较小。结果见表2。
2.2 含量测定
2.2.1 色谱条件 以Venusil XBP Polar-Phenyl(250 mm×4.6 mm,5 μm)为色谱柱,以甲醇-0.1%磷酸溶液(含0.04%三乙胺)(1.5 ∶ 98.5,V/V)为流动相;检测波长为210 nm;流速为1 mL/min;柱温为30 ℃;进样量为10 μL。
2.2.2 供试品溶液的制备 将用酸和碱溶液预处理过的阳离子交换树脂装入交换柱(直径2 cm,柱高20 cm),并经酸-碱-酸处理(用1 mol/L稀盐酸缓慢流过树脂,用量约为树脂体积的3倍,再用酸浸泡1 h后,用水冲洗至洗脱液pH为5左右;随后用1 mol/L氯化钠缓慢流过树脂,用量与酸相同,再用碱浸泡1 h后,用水洗至洗脱液pH为9左右;再以上述同样方法进行酸处理)后转型为H+型。取“2.1.2”项下越婢加术汤50 mL,用1 mol/L稀盐酸将越婢加术汤调节pH至1~2后,以每小时1.5倍柱体积流过已转型的交换柱,并用水冲洗至洗脱液完全无色,再用 2 mol/L氨水100 mL以每小时1.5倍柱体积洗脱,收集氨水洗脱液,于60 ℃水浴中蒸干,残渣用2 mL甲醇复溶,经0.22 μm微孔滤膜滤过,取续滤液,即得供试品溶液。
2.2.3 混合对照品溶液 精密称取盐酸麻黄碱、盐酸伪麻黄碱对照品,用甲醇溶解,制得盐酸麻黄碱、盐酸伪麻黄碱质量浓度分别为0.048、0.051 mg/mL的混合对照品溶液。
2.2.4 阴性对照溶液 按“2.1.2”项下方法制备缺麻黄的阴性对照药液,再按“2.2.2”项下方法制备阴性对照溶液。
2.2.5 系统适用性试验 取上述混合对照品溶液、供试品溶液(编号S1)和阴性对照溶液,按“2.2.1”项下色谱条件进样测定,记录色谱图。结果显示,盐酸麻黄碱和盐酸伪麻黄碱与相邻色谱峰的分离度均大于1.5,理论板数按盐酸麻黄碱峰计均大于3 000,阴性对照对两种成分的测定无干扰。结果见图4。
2.2.6 线性关系考察 取“2.2.3”项下混合对照品溶液适量,加甲醇释释并定容,制得盐酸麻黄碱质量浓度分别为0.98、2.4、4.8、12、24、48 μg/mL,盐酸伪麻黄碱质量浓度分别为1.02、2.55、5.1、12.75、25.5、51 μg/mL的混合系列对照品溶液。取上述系列溶液,按“2.2.1”项下色谱条件进样测定,记录峰面积。以各待测成分的质量浓度(X,μg/mL)为横坐标、对应峰面积(Y)为纵坐标进行线性回归。结果显示,盐酸麻黄碱的回归方程为Y=2.0×107X-1 959.1(r=0.999 9),盐酸伪麻黄碱的回归方程为Y=1.0×107X-3 263.7(r=0.999 2),表明盐酸麻黄碱、盐酸伪麻黄碱在各自的质量浓度范围内与峰面积成良好的线性关系。
2.2.7 精密度试验 取“2.2.3”项下混合对照品溶液,按“2.2.1”項下色谱条件进样测定6次,记录峰面积。结果显示,盐酸麻黄碱、盐酸伪麻黄碱峰面积的RSD分别为1.37%、1.63%(n=6),表明仪器精密度良好。
2.2.8 重复性试验 取“2.1.2”项下越婢加术汤(编号S1),共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品中两种成分的含量。结果显示,盐酸麻黄碱、盐酸伪麻黄碱含量的RSD分别为1.42%、1.79%(n=6),表明方法重复性良好。
2.2.9 稳定性试验 取“2.2.2”项下供试品溶液(编号S1),分别于室温下放置0、2、4、8、12、24 h时按“2.2.1”项下色谱条件进样测定,记录峰面积。结果显示,盐酸麻黄碱、盐酸伪麻黄碱峰面积的RSD分别为1.37%、1.21%(n=6),表明供试品于室温下放置24 h内稳定性良好。
2.2.10 加样回收率试验 取已知含量的样品(编号S1),每份1 mL,共9份,每3份各按已知含量的0.5、1、2倍加入单一对照品溶液(取盐酸麻黄碱、盐酸伪麻黄碱对照品适量,加甲醇溶解并稀释,制得盐酸麻黄碱、盐酸伪麻黄碱质量浓度分别为0.048、0.051 mg/mL的单一对照品溶液)适量,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果显示,盐酸麻黄碱、盐酸伪麻黄碱的平均加样回收率分别为105.67%、104.15%,RSD均小于3%(n=9),表明方法准确度良好。结果见表3。
2.2.11 样品含量测定 按“2.1.2”项下方法制备10批越婢加术汤,按“2.2.2”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定,记录峰面积并按标准曲线法计算样品中两种成分的含量。每个样品平行测定3次,结果见表4。
2.3 CA
以10批越婢加术汤HPLC指纹图谱的20个共有峰峰面积为变量,以平方欧氏距离为测度,采用SPSS 26.0软件进行CA。结果显示,当距离为10时,10批样品可聚为3类,其中S1聚为一类、S3聚为一类,其余聚為一类。这表明10批样品质量存在一定差异,推测可能与饮片的产地差异相关。结果见图5。
2.4 PCA
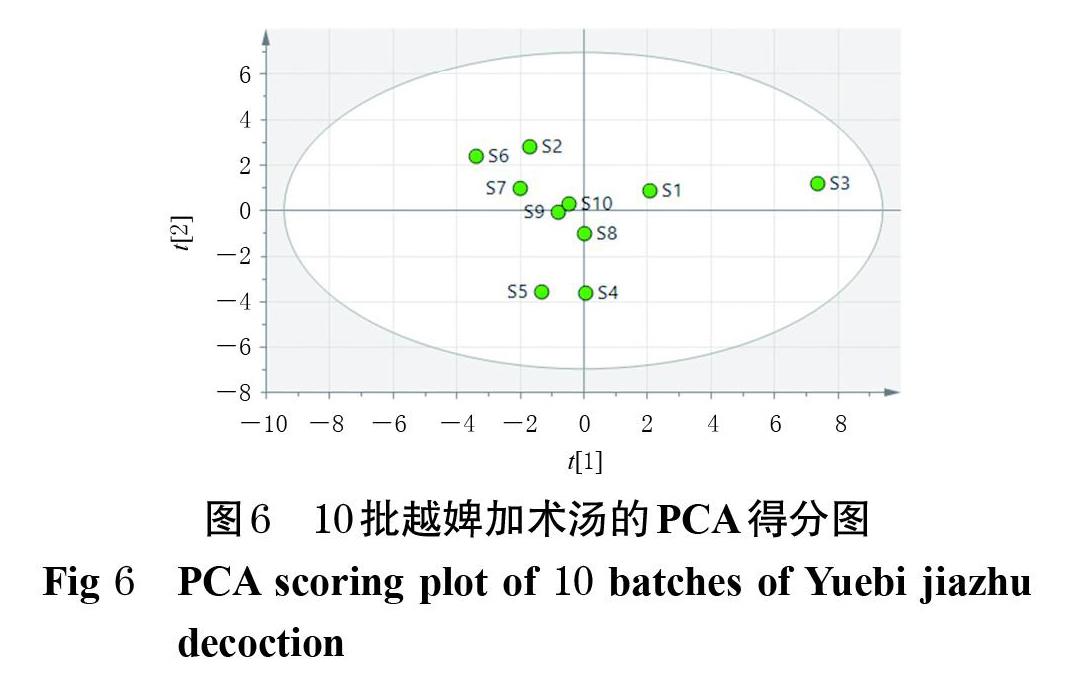
以10批越婢加术汤HPLC指纹图谱的20个共有峰峰面积建立10×20矩阵,采用SIMCA 13.0软件进行PCA。结果显示,S3位于得分图的最右侧,S1位于得分图的右侧,S2、S4~S10位于得分图的中部,与上述CA结果基本一致。结果见图6。
2.5 PLS-DA
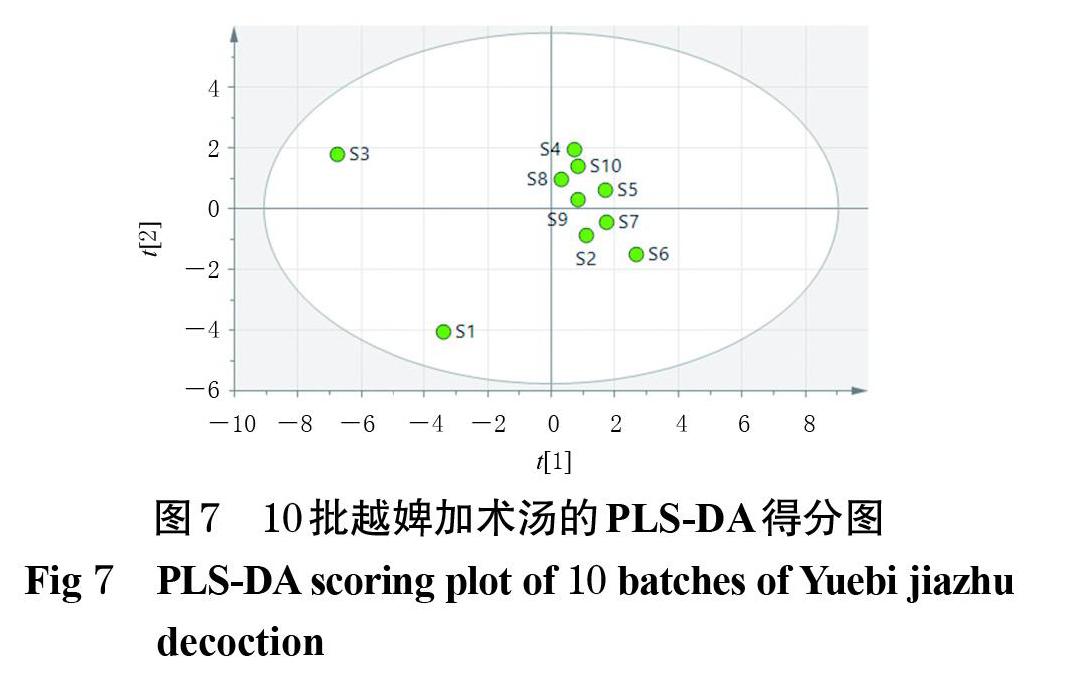
以10批越婢加术汤HPLC指纹图谱的20个共有峰峰面积建立10×20矩阵,采用SIMCA 13.0软件进行PLS- DA。由PLS-DA得分图可见,在95%置信区间内,10批样品聚成3类,其中S3位于得分图的左上侧,S1位于左下侧,S2、S4~S10位于中部偏右,与上述CA、PCA结果基本一致(图7)。在PLS-DA载荷图中,若数据点越偏离原点,表示其对应变量权重越大[22]。结果显示,20个共有峰中峰9、峰12、峰3、峰19的权重较大(图8)。
变量重要性投影(VIP)值越大,表明其对分类的贡献越大,以VIP值>1.0为标准[23],采用SIMCA 13.0软件筛选影响越婢加术汤质量的差异性成分。结果显示,有9个共有峰的VIP值>1,VIP值由大到小依次为峰9>峰3>峰12>峰8>峰19>峰18(甘草酸铵)>峰13>峰20>峰11(甘草苷)。结果见图9。
3 讨论
3.1 色谱条件的确定
3.1.1 指纹图谱的色谱条件 本课题组前期分别对Waters Atlantis T3(150 mm×4.6 mm,5 μm)、Waters XSelect HSS T3(150 mm×4.6 mm,5 μm)、Hypersil BDS C18(250 mm×4.6 mm,5 μm)等3种不同类型的C18色谱柱进行了考察,结果发现,采用封尾的Hypersil BDS C18时,所得色谱峰峰形对称、分离度好且基线平稳,故选择指纹图谱的色谱柱为Hypersil BDS C18。同时,本课题组对甲醇-0.1%甲酸溶液、乙腈-0.1%甲酸溶液、甲醇-0.1%磷酸溶液以及乙腈-0.1%磷酸溶液(含0.04%三乙胺)等不同流动相体系进行了比较,结果显示,前3种流动相体系所得色谱峰峰形较差且信息较少,故选择乙腈-0.1%磷酸溶液(含0.04%三乙胺)为流动相。此外,本课题组还比较了不同检测波长(210、230、250、270、290 nm)下的色谱图,结果显示,当检测波长为230 nm时,所得色谱峰数量较多,能较全面、完整地反映越婢加术汤整方的化学成分信息,虽然盐酸麻黄碱和盐酸伪麻黄碱在该波长条件下均不出峰,暂未能指认这两种成分,但鉴于中药指纹图谱的整体性,故选择检测波长为230 nm。
3.1.2 含量测定的色谱条件 本课题组前期分别对Waters Atlantis T3(150 mm×4.6 mm,5 μm)、Waters XSelect HSS T3(150 mm×4.6 mm,5 μm)、Hypersil BDS C18(250 mm×4.6 mm,5 μm)等3种不同类型的C18色谱柱进行了考察,结果发现,上述C18色谱柱所得盐酸麻黄碱和盐酸伪麻黄碱的色谱峰峰形较差,通过参照2020年版《中国药典》(一部)“麻黄”项下含量测定的色谱条件,本研究最终选择了麻黄专用柱(Venusil XBP Polar-Phenyl)[18]。同时,本课题组又对甲醇-0.1%磷酸溶液(含0.04%三乙胺)、甲醇-0.1%磷酸溶液(含0.1%三乙胺)以及甲醇-0.1%磷酸溶液(含0.2%三乙胺)等不同流动相体系进行了比较,结果发现,以甲醇-0.1%磷酸溶液(含0.04%三乙胺)为流动相时,所得色谱峰峰形较好。此外,本课题组还比较了不同检测波长(205、207、210、213、215 nm)下的色谱图,结果显示,当检测波长为210 nm时,柱效更高且峰形更好。
3.2 供试品处理方法的确定
本课题组前期比较了越婢加术汤未经处理、甲醇溶 解未经超声处理以及甲醇超声处理等3种方法,结果发现,以甲醇超声处理后所得样品色谱图的杂峰较少,有利于共有峰及待测成分色谱峰的分析与辨认。含量测定供试品处理方法主要包括有机溶剂萃取法、加酸超声法和阳离子交换树脂法等[24-25],本课题组前期参考上述文献,结果发现,经阳离子交换树脂法纯化后盐酸麻黄碱和盐酸伪麻黄碱能够完全分离,所得的色谱峰峰形好。
3.3 测定结果的分析
由于盐酸麻黄碱和盐酸伪麻黄碱结构的特殊性,无法与其他活性成分在同一波长下被检出,故为尽可能体现更多成分,在指纹图谱中暂未指认上述两种成分。指纹图谱结果显示,10批越婢加术汤的HPLC叠加指纹图谱共有20个共有峰,其与对照指纹图谱的相似度均大于0.92。经与混合对照品对比,指认峰11为甘草苷,峰18为甘草酸铵。
由于越婢加术汤中活性成分较多,其君药麻黄中的主要活性成分为盐酸麻黄碱和盐酸伪麻黄碱[6],并鉴于2020年版《中国药典》(一部)中“麻黄”项下含量测定的指标成分为盐酸麻黄碱和盐酸伪麻黄碱[18],故选择这两种成分为含量测定的指标。含量测定结果显示,10批越婢加术汤中盐酸麻黄碱和盐酸伪麻黄碱的含量分别为0.008 1~0.014 3、0.002 5~0.011 8 mg/mL,其中S4、S5样品中上述成分的含量相对较低。由于仅有S4样品中麻黄饮片的产地为河北,提示河北产麻黄饮片所含有效成分的含量与其他产地样品存在较大差异;而S5样品中的各味饮片有与其他批次饮片相同的产地,由此推测相同产地不同批次的麻黄饮片质量也存在差异。
3.4 化学模式识别结果的分析
CA、PCA、PLS-DA结果均显示,10批越婢加术汤可聚为3类,S1、S3分别为一类,其余为一类。这表明不同产地或相同产地不同批次饮片对越婢加术汤的质量均有影响,其中S3与其余样品的差异较为明显,将其所含饮片产地与其余批次进行比较后推测,新疆产甘草与其余产地产甘草的质量存在差异(S3样品中的各味饮片有与其他批次饮片相同的产地,仅甘草产地为新疆)。有9个共有峰的VIP值>1,分别为峰9、峰3、峰12、峰8、峰19、峰18(甘草酸铵)峰13、峰20、峰11(甘草苷),其中峰9、峰3、峰12、峰8对应成分的VIP值较高,经与各单味饮片的色谱图比较后发现,上述4个色谱峰均来自麻黄,提示麻黄可能是造成越婢加术汤质量差异的重要因素。
综上所述,本研究所建HPLC指纹图谱和含量测定方法操作简便、准确,结合化学模式识别可用于越婢加术汤的质量控制。峰9等9个成分为影响越婢加术汤质量的差异性成分。
参考文献
[ 1 ] 张仲景.金匮要略[M].北京:中国医药科技出版社,2018:45-47.
[ 2 ] 佟国莲,赵虹.越婢加术汤治疗风湿热痹的疗效观察[J/OL].临床医药文献电子杂志,2017,4(9):1721[2021-07-10]. https://d.wanfangdata.com.cn/periodical/lcyydzzz201709098.DOI:10.3877/j.issn.2095-8242.2017.09.098.
[ 3 ] 刘文艳,韩健,梁丽梅,等.越婢加术汤的急性毒性实验[J].中国老年学杂志,2013,33(6):1330-1331.
[ 4 ] 但文超,丁宇坤,何庆勇.越婢加术汤临床应用经验浅析[J].北京中医药,2019,38(10):1018-1020.
[ 5 ] 郑腾飞,胡蓝方.越婢加术汤临床证治体悟[J].世界最新医学信息文摘,2019,19(30):265.
[ 6 ] 卓小玉,陈晶,田明,等.麻黄的化学成分与药理作用研究进展[J].中医药信息,2021,38(2):80-83.
[ 7 ] 左军,张金龙,胡晓阳.白术的化学成分及现代药理作用研究进展[J/OL].辽宁中医药大学学报,2021,23(10):6-9 [2021-07-10]. http://kns.cnki.net/kcms/detail/21.1543.R.20210419.1116.007.html. DOI:10.13194/j.issn.1673-842x.
2021.10.002.
[ 8 ] 邓桃妹,彭灿,彭代银,等.甘草化学成分和药理作用研究进展及质量标志物的探讨[J].中国中药杂志,2021,46(11):2660-2676.
[ 9 ] 赵文竹,张瑞雪,于志鹏,等.生姜的化学成分及生物活性研究进展[J].食品工业科技,2016,37(11):383-389.
[10] 王远,邢丽杰,张昊伟,等.红枣中活性成分及其分析方法研究进展[J].农产品加工,2021(4):62-66.
[11] 李园,程悦,朋汤义.浅析影响中药临床療效的因素及应用措施[J].陕西中医药大学学报,2021,44(5):54-58.
[12] 付会玲.越婢加术汤合五苓散治疗成人原发性肾病综合征风水相搏证临床观察[J].四川中医,2020,38(2):125-128.
[13] 俞惠英,路新强.越婢加术汤治疗风湿热痹37例疗效观察[J].亚太传统医药,2013,9(6):152-153.
[14] 沈晗,黄玉宇,沈夕坤,等.五味消毒饮HPLC指纹图谱建立及10种成分同时测定[J].中成药,2021,43(1):22-28.
[15] 李海伦,李恒,孙飞,等.经典名方大秦艽汤HPLC指纹图谱及含量测定方法研究[J].中草药,2021,52(1):99-107.
[16] 刘鹤,贺丹彤,邢春来,等.甘姜苓术汤HPLC指纹图谱及含量测定方法的建立[J].药物分析杂志,2021,41(1):42-50.
[17] 孙立丽,王萌,任晓亮.化学模式识别方法在中药质量控制研究中的应用进展[J].中草药,2017,48(20):4339- 4345.
[18] 国家药典委员会.中华人民共和国药典:一部[S]. 2020年版.北京:中国医药科技出版社,2020:333-334.
[19] 李明,杨丽娜.基于中医汤剂的中药常用剂量研究进展[J].中国实验方剂学杂志,2018,24(8):12-16.
[20] 范丽丽,邓家刚,郝二伟.中药剂量与功效相关性研究进展[J].中国中医药信息杂志,2010,17(10):104-106.
[21] 高晓山,王旭华,陈馥馨.大枣剂量的数量与衡量讨论[J].中国中药杂志,2006,31(22):1909-1911.
[22] 靳贝贝,裴香萍,梁惠珍.青皮药材的HPLC指纹图谱建立及聚类分析和主成分分析[J].中国药房,2018,29(24):3336-3339.
[23] 薛起梅,施海蔚,钱叶飞,等.基于HPLC指纹图谱结合化学模式识别的黄蛭益肾胶囊质量控制[J].药物评价研究,2021,44(5):956-963.
[24] 雍子宜,张旭,高秀容,等.固相萃取高效液相法测定复方川贝精胶囊中3种麻黄碱成分的含量[J].中国药师,2020,23(5):1003-1005.
[25] 唐乔. HPLC法测定百咳静颗粒中8个成分的含量[J].药物分析杂志,2021,41(1):147-153.
(收稿日期:2021-05-28 修回日期:2021-10-13)
(编辑:陈 宏)
