PCDH15基因变异相关耳聋患者基因型与听力表型特征分析
2021-12-10施韬关静李进赵翠兰兰王大勇王洪阳王秋菊
施韬关静李进赵翠 兰兰王大勇 王洪阳王秋菊*
1 浙江中医药大学医学技术与信息工程学院(杭州 310053)
2 中国人民解放军总医院耳鼻咽喉头颈外科医学部,国家耳鼻咽喉疾病临床医学研究中心,解放军耳鼻咽喉研究所,聋病教育部重点实验室;聋病防治北京市重点实验室(北京 100853)
PCDH15变异导致Usher综合征1型(USH1)或常染色体隐性遗传性耳聋23型(DFNB23)。USH1是一种以进行性视网膜色素变性(retinitis pig‐mentosa,RP)和先天性感音神经性听力损失为特征的常染色体隐性疾病,伴有先天性前庭功能障碍,运动发育迟缓、难以保持平衡[1-4]。USH1在视力异常表型出现之前,与非综合征型耳聋(Non-syn‐dromic hearing loss,NSHL)表型相同,也被称为拟非综合征型耳聋(mimics NSHL)[5]。因此,当临床中发现PCDH15变异的患者,需监测随访以鉴别是否可能为USH1。
目前,已报道的PCDH15变异位点为144个(http://www.hgmd.cf.ac.uk/,Update 2020/04),大多为国外报道,且很少有针对PCDH15变异患者的描述,国内相关报道更少。本研究通过新一代测序技术,在4个家系中检出PCDH15复合杂合变异,并分析4个家系的基因型及表型特征。
1 研究对象
研究对象为2016年3月-2019年6月在解放军总医院耳鼻咽喉头颈外科就诊的4个汉族耳聋家系(图1),共18人,6名患者均为先天性极重度感音神经性耳聋,男性2名、女性4名,分别来自河北、北京、河南、山东;其中3名儿童,3名成人。收集先证者及其父母或家属血样,共18份。所有研究对象均已签署知情同意书,未成年子女由其父母签署同意书。

图1 PCDH15变异相关耳聋患者家系及听力图。B1:+/+表示c.3441 del A/c.2528 C>A;A2:患者9月左侧植入CI后助听听阈;B2:患者29岁双耳听阈;C2:患者11月双耳听阈;D2:患者10月龄右侧植入CI后双耳听阈。Fig.1 Pedigrees of deafness families related to variants of PCDH15.B1:+/+mean by c.3441 del A/c.2528 C>A;A2:The patient’s left hearing threshold after left cochlear implant in 9 months ago;B2:The patient’s binaural hearing threshold in 29 years old;C2:The patient’s binaural hearing threshold in 11 months old;D2:The patient’s binaural hearing threshold after right cochlear implant in 10 months ago.
2 研究方法
2.1 临床资料收集
采用项目组自主设计的问卷调查表,记录患者病史,完成家系调查并绘制家系图。对患者进行听力学检查,包括纯音测听、声导抗、畸变产物耳声发射(distortion product otoacoustic emissions,DPOAE),对婴幼儿进行听性脑干反应(auditory brainstem re‐sponse,ABR)、40Hz听觉事件相关电位(40 Hz au‐ditory event related potential,40Hz AERP)及听觉稳态反应(auditory steady state responses,ASSR)检查。听力损失程度根据500、1k、2k、4kHz平均听阈分类,分为26-40dB HL轻度、41-55 dB HL中度、56-70 dB HL中重度、71-90 dB HL重度以及>90 dB HL极重度听力损失。对患者进行眼科检查及影像学检查,包括眼底镜检查、颞骨CT、内听道MRI,评估患者眼部是否存在异常。对患者进行1-3年电话随访及复诊,对迟发性眼部临床表型进行长期的关注。
2.2 基因测序
采集的血样提取基因组DNA,试剂盒由天根生物科技(北京,中国)提供。将DNA片段化及末端修复之后,构建DNA文库。运用新一代测序技术,对其中2名患者进行目标区域测序(Targeted Regions Sequencing,TES),2名患者全外显子测序(Whole exome sequencing,WES)。通过生物信息分析方法,确定可疑致病基因变异,先证者及其亲属均进行变异位点的Sanger验证。
2.3 致病性分析
得到变异位点后,根据美国医学遗传学与基因组学会(The American College of Medical Genetics and Genomics,ACMG)指南将变异分为致病变异、疑似致病变异、意义不明的变异以及良性变异[6-8]。对于拷贝体变异(Copy number variations,CNVs),同样参考ACMG指南[9]。
3 研究结果
3.1 听力临床表型
1707699 (图1-A1:III.1),女,8岁,2月龄ABR检测,双耳气骨导在100dB nHL均无反应;ASSR检测,双耳100dB nHL均无反应;40Hz AERP检测,双耳100 dB nHL均无反应。
1707867 (图1-B1:II.3),女,29岁,先天性聋,右耳平均听阈96.25dB HL,左耳平均听阈103.75 dB HL,声导抗正常。II.2先天性聋,右耳平均听阈105dB HL,左耳平均听阈107.5 dB HL。
1808120 (图1-C1:III.3),男,3岁,7月龄ABR检测,双耳气导在100dB nHL均无反应;ASSR检测,双耳100dB nHL均无反应;DPOAE检测,双耳各频率均未引出;声导抗显示双耳A型。
1908312 (图1-D1:II.1),女,4岁,10月龄ABR检测,双耳气导在95dB nHL均无反应;ASSR检测,双耳平均听阈均为116dB nHL;DPOAE检测,双耳各频率均未引出。患儿于10月龄右侧植入人工耳蜗(cochlear implant,CI),助听后右耳平均听阈23.75dB HL。
四名先证者颞骨CT检查正常,提示无内耳畸形。
3.2 基因检测结果
家系1707699和1908312的先证者及父母进行WES检测,家系1707867和1808120的先证者进行TES检测,共检测出7种PCDH15变异(NM_033056.3),2种无义变异,2种剪接位点变异,1种错义变异,1种移码变异,1种CNV(表1)。7种变异中,c.733C>T为已报道的致病变异位点[2],c.2528C>A为已报道的意义不明的变异位点[10],根据该家系表型及ACMG指南建议(PM2+PP1+PP3,PM表示证据强度中等,PP表示辅助证据),考虑该变异位点为疑似致病。其余均为新发现的变异。

表1 基因检测结果及分析Table 1 Summary and analysis of genotype
对先证者父母及家族成员(12人)进行Sanger验证,结果显示所有亲属均为PCDH15变异单杂合携带者。家系1707699中,III.1为EX19-21del和c.1997+1G>A复合变异遗传模式,EX19-21长度约为27kb,III.2检测结果为EX19-21 del单杂合携带。家系1707867中,II.3为c.3441 del A和c.2528C>A复合杂合变异,II.1和II.2与II.3的变异位点相同。家系1808120中,II.3为c.733C>T和c.157+1G>A,家系1908312中,II.1为c.733C>T和c.2756del T,均携带c.733C>T这个变异位点。所有先证者均符合常染色体隐性遗传模式。
3.3 随访结果
得到变异位点后,为明确患者病因,需对患者及携带者进行随访,告知有可能存在迟发性RP表型的可能性,强调视力方面检查的重要性(表2)。

表2 先证者表型汇总Table 2 Summary of phenotype of probands
1707699 (图1-A1:III.1)于9月龄时左侧植入CI,植入后左耳平均听阈为41.25dB HL,3岁时为31.25dB HL(图2-B)。双音节言语识别率为100%,语句识别为95%。患儿18月龄能独立行走,双眼远视、散光,5岁时眼底镜检查正常(图2-A)。III.2为EX19-21del杂合携带者,目前三岁,听力正常,言语发育可。

图2 170769眼底镜检查与助听后听阈变化。A为患者眼底镜检查结果(正常);B为患者植入CI听阈变化。Fig.2 Ophthalmofundoscopy and threshold change after CI of 170769.A:result of ophthalmofundoscopy(normal);B:threshold change after CI.
1707867 (图1-B1:II.3),主诉无眩晕、走路不稳等前庭相关功能异常,视力在正常范围之内。II.1和II.2临床表型与其相同,II.1目前33岁,13岁时右侧植入CI,使用两年后效果不佳,后未再佩戴。II.2和II.3均双侧佩戴助听器,自述交流能力一般。III代子女听力均正常,视力在正常范围之内,年龄10月-13岁。
1808120 (图1-C1:III.3),11月龄左侧植入CI,交流能力尚可。患儿确诊为运动发育迟缓,走路和下楼梯经常性的摔倒。由于年龄限制,无法进行视网膜电图检查,家长拒绝进行眼底镜检查。
1908312 (图1-D1:II.1),10月龄右侧植入CI,交流能力尚可。18月龄独立行走,视力双眼远视及散光,存在夜晚外出困难的症状,由于年龄限制,无法进行视网膜电图检查,家长拒绝进行眼底镜检查。
4 讨论
PCDH15与钙粘蛋白23是内耳毛细胞静纤毛顶连接(tip-link)的重要组成部分,内耳毛细胞是一种机械-电传感器,完成声电转换,帮助产生听觉和平衡知觉[11,12]。毛细胞顶端存在静纤毛,静纤毛的摆动可以通过tip-link的控制打开机械门控通道[13,14]。PCDH15变异改变蛋白结构域的空间形态,机械门控通道出现异常,引起USH1F或DF‐NB23[1,2,1517]。Alagramam等在成人和胎儿视网膜及内耳内发现PCDH15的表达[1]。Ahmed等发现在人眼的感光细胞中存在PCDH15,这表明USH1患者在视力方面存在缺陷,将出现进行性RP[16]。因此,USH1临床表型包括耳聋、前庭功能障碍和RP。

表3 在不同种族中PCDH15导致的USH1Table 3 USH1 caused by PCDH15 in different races
PCDH15变异导致的USH1是一种典型的mimics NSHL,患者在早期仅表现出耳聋特征,而RP是迟发性的。通常,患者10岁左右将会出现夜盲和隧道视觉等RP早期症状,随后视野进行性向心性缩小,直至完全失明[18]。有研究发现,患者在21岁时才出现RP早期症状[3]。1908312已出现夜盲的症状,并且其与1707699均为远视及散光。有研究表明在NSHL儿童中USH1发生率为1.3-2.2%[22]。在Vonam等的研究中,新一代测序能使USH1的诊断年龄早于视力损失的发病年龄[24],为USH1家庭带来许多即时和长期的帮助[17]。本研究通过基因检测后的随访,3名最初诊断为NSHL的儿童,在检测出PCDH15变异后,随访发现视力及前庭的异常表型。
PCDH15是导致USH1的主要致病基因,在德系犹太人中PCDH15是最常见的致病基因,c.733C>T在该人群中存在始祖效应,占据USH1的比例为50%-60%,c.733C>T的携带率为0.79%-2.48%[2,20]。在本研究中,2名患者均有c.733C>T致病位点,考虑将其作为中国人群PCDH15热点变异进行下一步研究。
PCDH15变异导致的USH1,除了可以通过单核苷酸变异致病,CNV同样也是关键致病变异类型。CNV与遗传性耳聋关系密切[25]。1707699外显子19-21缺失,长度约为27kb,CNV是指大于1kb的DNA片段的缺失/重复[26]。大多数已报道的CNV集中在PCDH15前5个外显子及第20外显子附近,在本研究中检测出的EX19-21 del包含在已报道的CNV范围内(表4)。由此可见,PCDH15基因除了点变异,CNV也是的主要变异类型。
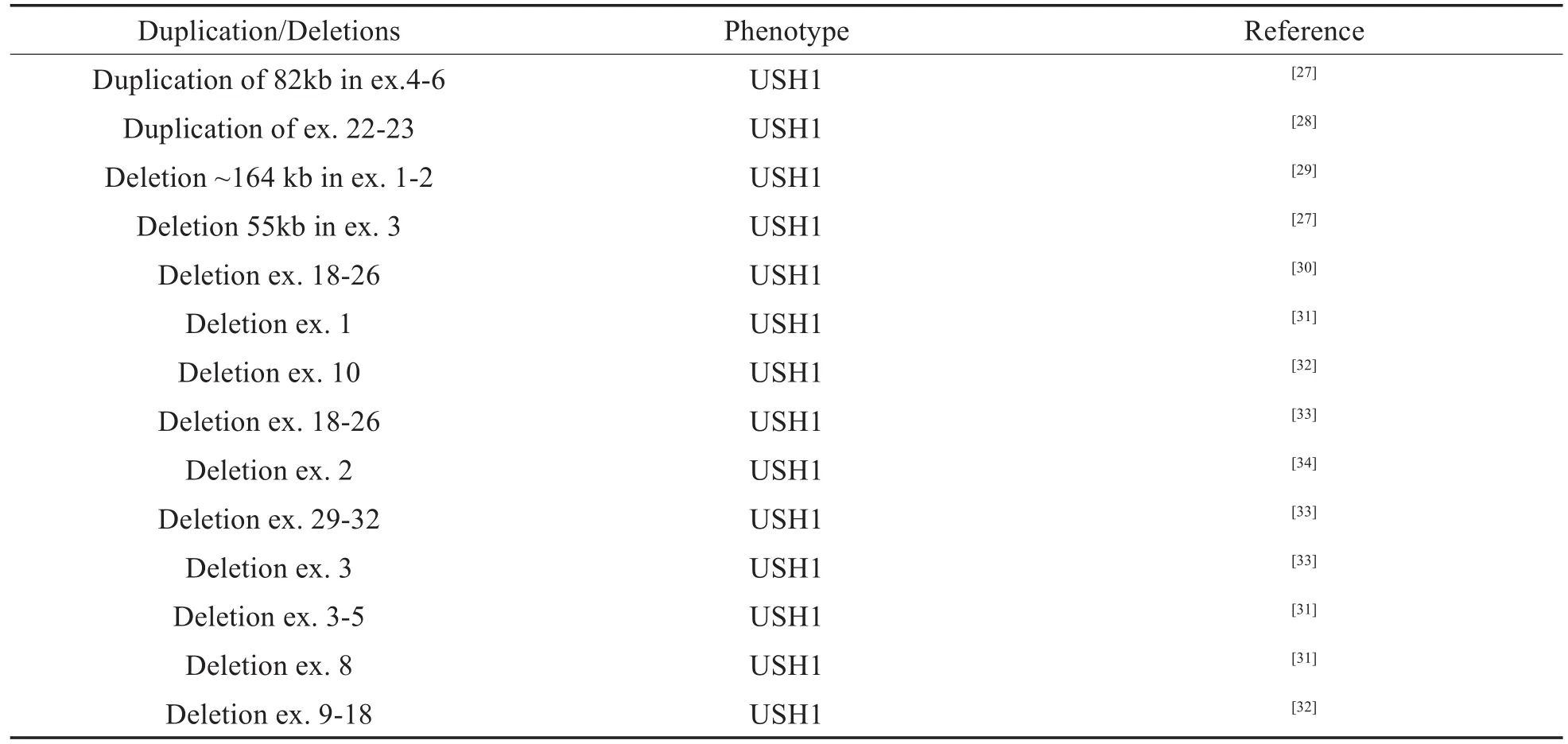
表4 PCDH15已报道的拷贝体变异Table 4 CNVs has been reported in PCDH15
PCDH15表达于毛细胞静纤毛间的tip-link,其变异影响耳蜗的机电转换,CI是通过手术,将电极片植入内耳,直接作用于螺旋神经节细胞,绕过耳蜗的机电转换,进而帮助患者感知声音[35-37]。在本次研究中,通过随访发现,3例儿童CI植入效果较好,1707699植入后双音节词言语识别率达到100%。1707867的哥哥在13周岁时才植入CI,效果不佳,佩戴两年后未再使用。通过这些发现,PCDH15变异的耳聋患者均存在先天性重-极重度听力损失,早期植入CI,可以让儿童在言语发育关键期接受更多的声音刺激,言语发育更好,后期儿童可以通过视力阅读唇语,配合听觉康复来获得更好地言语发展[2-3,38]。
PCDH15变异是导致DFNB23和USH1的重要原因,USH1在临床上表现为双耳先天性重-极重度听力损失、运动发育迟缓及进行性RP。本研究为4个极重度感音神经性聋家系明确了分子病因,发现均为PCDH15复合杂合变异,其中有7个变异位点,包括2种无义变异,2种剪接位点变异,1种错义变异,1种移码变异,1种CNV,5个为新发变异位点;考虑c.733C>T为热点变异位点,需进一步研究;通过随访,分析PCDH15致病患者听力、视力、前庭功能的临床表型,及CI佩戴效果,发现对于PCDH15基因变异导致的先天性极重度聋的患儿,在早期植入CI言语发育尚可,为PCDH15变异导致的USH1及DFNB23的分子诊断及治疗干预提供参考意见,USH1作为mimics NSHL的一种,早期诊断及干预至关重要。
