对二甲苯分子在激发过程中分子构型和简谐振动频率的变化
2021-12-01张泽霞沈丽娜
秦 晨 张泽霞 沈丽娜
(新疆师范大学物理与电子工程学院,新疆 乌鲁木齐 830054)
0 引言
对二甲苯(p-Xylene,PX)分子是一种重要的有机化工原料,是生产对苯二甲酸、涤纶纤维、医药和农药的重要原料。PX分子广泛存在于各种环境介质中,有一定的毒性,对PX分子的激发态构型和振动情况进行研究,可以深入了解该分子的光化学和光物理性质,为进一步消除环境介质中残留的PX污染物分子提供研究基础。
近年来,对PX分子的研究主要集中在各种介质中痕量PX的污染检测、生物降解[1]、催化氧化及制备技术和外场效应[2]、激发态以及离子态光谱等方面[3],杜建宾等人[2]利用DFT的方法对PX分子在不同电场下的振动频率和IR光谱进行了研究。目前还没有对PX分子在基态(S0态)和第一电子激发态(S1态)的所有简谐振动模式以及激发过程中分子构型变化的研究。
该文通过计算得到了PX分子在S0和S1态的键长、键角和简谐振动频率,并结合Wilson标识法对所有振动频率的振动模式进行标识。通过分析得到了PX分子在S1←S0跃迁过程中几何构型的变化和振动频率的变化情况,进一步了解了对位的2个甲基(CH3)取代基对PX分子激发态构型变化、电子态跃迁和简谐振动情况等的影响,这些研究结果对理解PX分子在紫外光(UV)作用下的光化学和光物理过程有十分重要的作用。
1 理论计算
PX分子的分子式为C8H10,共有18个原子,其分子结构及原子标号如图1所示。图1中的1、2、3、4、5、6、11和15号原子表示C原子,其余标号的原子是H原子。利用Gaussian 16软件的从头算法(ab initio)和密度泛函理论(DFT)方法预测了PX分子在S0态和S1态优化的分子构型和简谐振动频率,进而得到该分子在激发过程中的几何构型和振动频率的变化,对同一振动模式对应振动频率在激发过程中的变化情况进行了讨论。PX分子在S0态的几何结构和振动频率可以利用RHF、RB3LYP和RB3PW91方法在6-31G基组水平上进行模拟计算。PX分子在S1态的几何结构和振动频率可以用单组态相关CIS、TD-B3LYP和TD-B3PW91方法在6-31G基组水平上进行模拟计算。
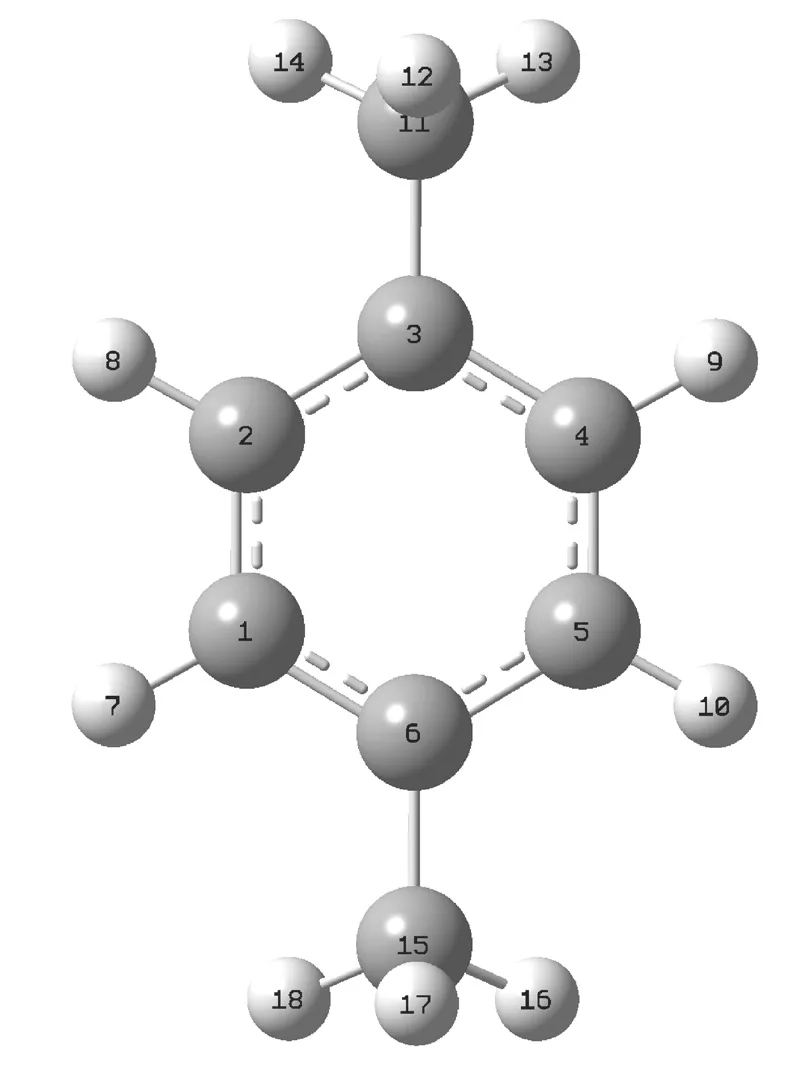
图1 PX分子的结构示意图及原子标号
2 结果与讨论
2.1 PX分子在S1←S0的跃迁过程中分子构型的变化
表1给出了分别利用RB3LYP和TD-B3LYP的方法在6-31G基组水平上得到的PX分子S0和S1态优化的几何构型参数。由于2个CH3取代基取代苯环上的H原子后会分别向苯环贡献电子,因此会改变苯环上的电子密度分布,进而改变分子的几何结构。通过分析PX分子在S1←S0激发过程中键长和键角的变化情况,可以得到该分子在该激发过程中分子构型的变化情况。

表1 PX分子在S0和S1态优化的键长和键角。
由表1可以看到,在S1←S0的跃迁中,键长C1-C2、C2-C3、C3-C4、C4-C5、C5-C6和C6-C1分别从1.39690 Å、1.40169 Å、1.40193 Å、1.39665 Å、1.40193 Å和1.40169 Å增大到1.42379 Å、1.43032 Å、1.43033 Å、1.42380 Å、1.43033 Å和1.43033 Å,CH3上的C-H键的平均键长从1.09580 Å增大到1.09857 Å,苯环上的C-H键的平均键长从1.08769 Å减小到1.08507 Å,这些键长的变化说明π*←π的激发使苯环发生了扩张。值得注意的是键长C3-C11和C6-C15均从1.51199 Å减少到了1.49246 Å,这2个键长变短就说明CH3与苯环的相互作用在S1态时比在S0态时强。另外在S1←S0的跃迁中,键角∠C2C3C4和∠C5C6C1均从S0态的117.6º变为S1态的119.6º,键角的增加也说明了CH3与苯环的相互作用在S1态时比在S0态时强。
2.2 PX分子在S1←S0跃迁过程中振动模式的变化
因为PX分子共有18个原子,所以有48个简正振动模式,包括30个苯环上的振动模式和18个CH3的振动模式。表2给出了利用RB3LYP和TD-B3LYP方法在6-31G基组水平上进行模拟计算得到的PX分子在S0和S1态的振动频率,并结合Wilson标识法对模拟计算得到的48个振动频率进行振动模式标识。
表2中PX分子的简谐振动一共有48种,相关描述如下。
PX分子的苯环所在平面外弯曲振动模式共有9种,用符号γ来表示,见表2。在S0态,模拟计算得到的频率值 为137 cm-1、418 cm-1、310 cm-1、497 cm-1、710 cm-1、850 cm-1、809 cm-1、974 cm-1和953 cm-1的振动频率,分别被标识为16b1、16a1、41、111、10b1、10a1、17b1、17a1和51振动模式(a和b为振动模式的符号)。在S1态,模拟计算得到的频率值为106 cm-1、201 cm-1、209 cm-1、439 cm-1、504 cm-1、539 cm-1、639 cm-1、652 cm-1和722 cm-1的振动频率,分别被标识为16b1、16a1、41、111、10b1、10a1、17b1、17a1和51振动模式。其中,振动模式16b1、16a1和41主要是苯环上的C-C-C键的苯环所在平面外的弯曲振动,振动模式111、10b1、10a1、17b1、17a1和51主要是苯环上的C-H和C-CH3的苯环所在平面外的弯曲振动。
PX分子的苯环所在平面的内弯曲振动模式共有10种,用符号β来表示,见表2。在S0态,模拟计算得到的频率值为461 cm-1、657 cm-1、836 cm-1、1 036 cm-1、1 341 cm-1、1 440 cm-1、1 339 cm-1、1 493 cm-1、1 620 cm-1和1 664 cm-1的振动频率,分别被标识为6a1、6b1、11、121、31、18b1、151、18a1、9b1和9a1振动模式。在S1态,模拟计算得到的频率值为410 cm-1、563 cm-1、805 cm-1、987 cm-1、1 317 cm-1、1 365 cm-1、1 444 cm-1、1 469 cm-1、1 507 cm-1和1 598 cm-1的振动频率,分别被标识为6a1、6b1、11、121、31、18b1、151、18a1、9b1和9a1振动模式。其中,振动模式6a1、6b1、11和121主要是苯环上的C-C-C键在苯环所在平面内的弯曲振动,振动模式31、18b1、151、18a1、9b1和9a1主要是苯环上的C-H和C-CH3在苯环所在平面内的弯曲振动。
PX分子的苯环所在平面内伸缩振动模式共有11种,用符号ν来表示,见表2。在S0态,模拟计算得到的频率值为386 cm-1、730 cm-1、990 cm-1、1 143 cm-1、1 210 cm-1、1 226 cm-1、1 238 cm-1、3 165 cm-1、3 164 cm-1、3 180 cm-1和3 184 cm-1的振动频率,分别被标识为8b1、131、19b1、141、8a1、7a1、19a1、7b1、20a1、20b1和21振动模式。在S1态,模拟计算得到的频率值为377 cm-1、712 cm-1、951 cm-1、1 106 cm-1、1 184 cm-1、1 214 cm-1、1 227 cm-1、3 194 cm-1、3 198 cm-1、3 210 cm-1和3 215 cm-1的振动频率,分别被标识为8b1、131、19b1、141、8a1、7a1、19a1、7b1、20a1、20b1和21振动模式。其中,振动模式8b1、19b1、141、8a1和19a1主要是苯环上的C-C键在苯环所在平面内的伸缩振动,振动模式131、7a1、7b1、20a1、20b1和21主要是苯环上的C-H和C-CH3在苯环所在平面内的伸缩振动。
PX分子的2个CH3取代基还有18种简谐振动模式,包括2个CH3转动、1个CH3面内弯曲振动、9个CH3上的C-H弯曲振动和6个CH3上的C-H伸缩振动,见表2。在S0态为52 cm-1和37 cm-1的振动频率以及在S1态为40 cm-1和80 cm-1的振动频率,分别被标识为在S0态和S1态的CH3转动。在S0态为286 cm-1的振动频率以及在S1态为289 cm-1的振动频率,被标识为在S0态和S1态的CH3面内的弯曲振动。在S0态为1 060 cm-1、1 066 cm-1、1 025 cm-1、1 420 cm-1、1 419 cm-1、1 491 cm-1、1 494 cm-1、1 504 cm-1和1 553 cm-1的振动频率以及在S1态为998 cm-1、1 000 cm-1、1 003 cm-1、1 393 cm-1、1 394 cm-1、1 445 cm-1、1 476 cm-1、1 481 cm-1和1 485 cm-1的振动频率,分别被标识为在S0态和S1态的CH3上的C-H弯曲振动。在S0态3 028 cm-1~3 114 cm-1的振动频率以及在S1态2 966 cm-1~3 099 cm-1的振动频率,分别被标识为在S0态和S1态的CH3上的C-H伸缩振动。
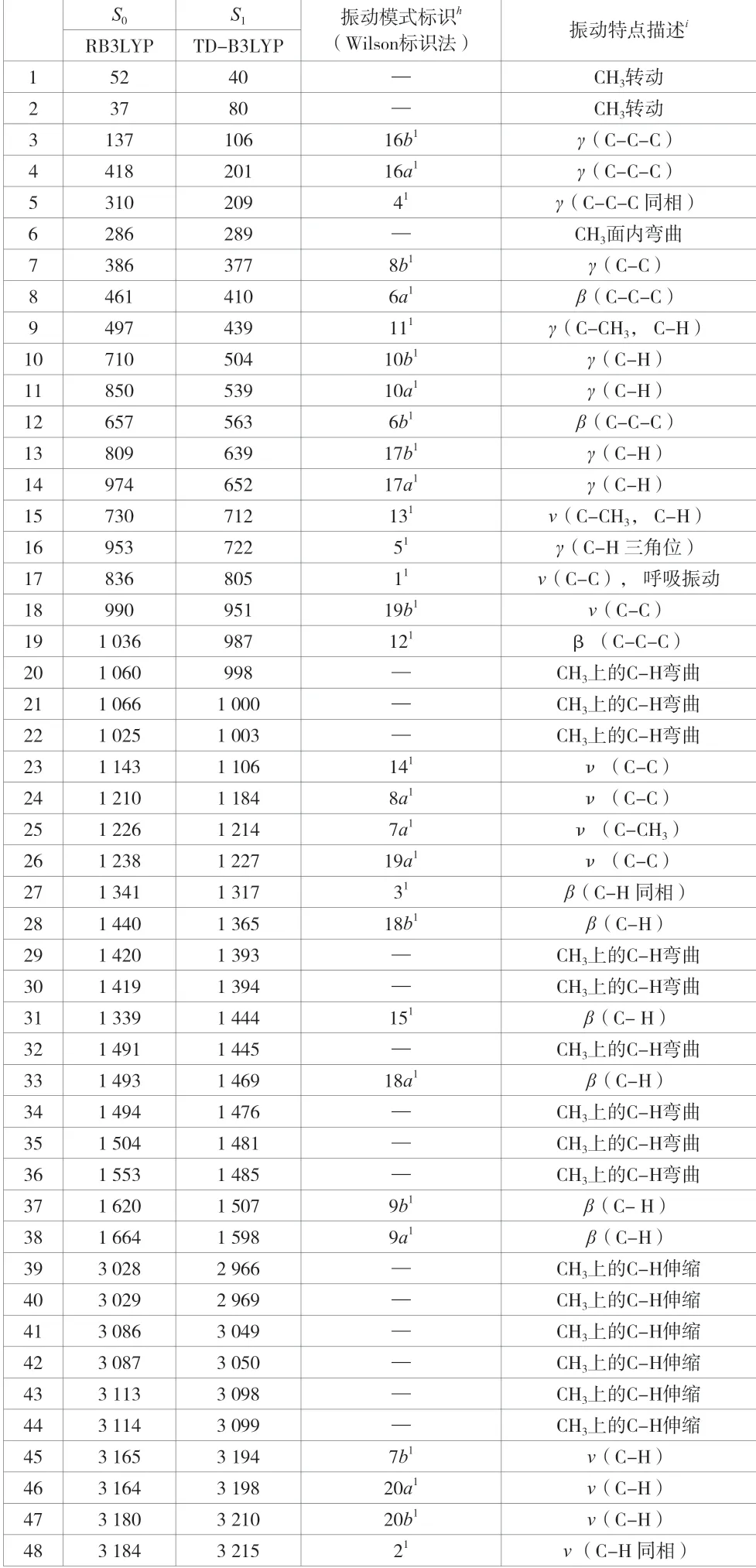
表2 PX分子在S0和S1态的振动频率值(单位:cm-1)和振动模式标识
综合上述标识结果和振动情况的描述可以看出,在S1←S0的激发过程中,PX分子的每种振动模式在S1态的频率值普遍低于其在S0态时的频率值,表明该分子在激发过程中结构发生了变化,苯环发生了扩张,结构变得松散了,也说明PX分子的结构在S0态的刚性强于在S1态的刚性。
PX分子的48种简谐振动模式中,那些在S1态有较大的Franck-Condon重叠的振动模式可以在振动光谱中被观察到。张嵩等人[3]用共振增强多光子电离给出了PX分子S1←S0的跃迁光谱图,同时用Gaussian软件的CIS/6-31G**方法预测了PX分析在S1态光谱图中的振动谱峰对应的理论值。表3列出了PX分子S1态的振动模式9b1、6a1、6b1、11、9a1和7a1对应的振动频率的实验值和理论值[3]以及该文用RB3LYP/6-31G方法给出的振动频率值。9b1、6a1、6b1、11、9a1和7a1的振动模式在实验谱图中给出的振动频率分别是369 cm-1、424 cm-1、553 cm-1、801 cm-1、937 cm-1和1 186 cm-1,用CIS/6-31G**的方法得到的振动频率分别是388 cm-1、433 cm-1、609 cm-1、851 cm-1、957 cm-1和1 297 cm-1,该 文 选 用 的RB3LYP/6-31G方法计算出来的振动频率分别是1 507 cm-1、410 cm-1、563 cm-1、805 cm-1、1 598 cm-1和1214 cm-1。通过对比可以发现,用RB3LYP/6-31G得到的振动模式6a1、6b1、11和7a1的频率值比用CIS/6-31G**的方法得到的频率值更加接近实验值,并且与实验中相应的频率值也非常接近。这里需要注意的是,用RB3LYP/6-31G得到的振动模式9a1和9b1的频率值与用CIS/6-31G**的方法得到的频率值相差较大。由于6a1、6b1和11的频率值在3种方法下的对比已经证实了RB3LYP/6-31G方法得到的频率值比用CIS/6-31G**方法得到的频率值更加接近实验结果,因此,这里振动模式9a1和9b1在选用不同计算方法时存在的差异可以解释为不同的作者对振动模式9a1和9b1给予了不同的标识的结果。由于计算过程中基组选择的不完备性,计算过程中忽略了电子相关性和振动的非简谐性,因此,不管哪种计算方法的计算结果都会略高于实验结果。考虑这些综合误差,给该文计算得到的6a1、6b1、11和7a1的频率值分别乘以一定的校正因子之后,计算结果与实验结果非常接近,两者吻合得非常好。注:f为参考文献[3]中实验的振动频率值和用CIS/6-31G**得到的振动频率值;g为该文中用TD-B3LYP/6-31G方法得到的振动频率值。
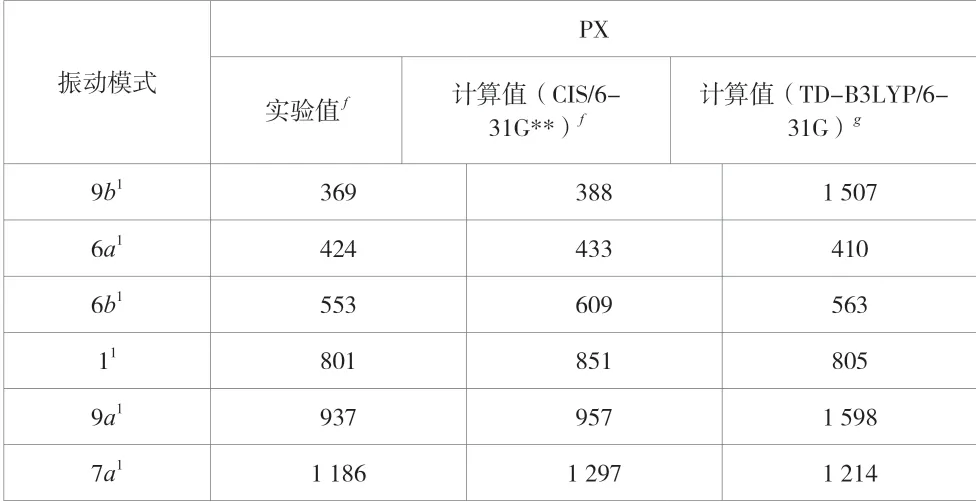
表3 对比PX分子S1态时同一振动模式对应的频率值(单位:cm-1)。
3 结论
对PX分子在S0态和S1态的键长、键角以及所有简谐振动频率值的分析分别选用的是利用RB3LYP方法和TDB3LYP方法得到的计算结果,标识振动模式时用到的是Wilson标识法。通过分析PX分子在S1←S0的跃迁过程中键长和键角的变化情况可以看到,在S1←S0的跃迁过程中π*←π的激发使PX分子的苯环发生了扩张,苯环的结构变得松散,CH3与苯环的相互作用在S1←S0的跃迁中发生了变化。同时,在S1←S0激发过程中,PX分子的每种振动模式在S1态的频率值普遍低于其在S0态时的频率值,说明PX分子的结构在S0态的刚性强于在S1态的刚性,同时CH3与苯环的相互作用在S1←S0的跃迁中也发生了变化。
