Mo-Bi-S@CNTs纳米双金属硫化物电催化CO2还原制取甲醇的性能
2021-11-25李春慧段东红张忠林卫国强刘世斌
池 琛,李春慧,段东红,张忠林,卫国强,李 瑜,刘世斌
(太原理工大学 化工学院,太原 030024)
二氧化碳(CO2)通常状态下是一种分子结构稳定、化学惰性的气体,其碳氧键键长为116 pm,介于碳氧双键(124 pm)和碳氧三键(113 pm)之间。近年来,煤炭、石油等化石燃料的大量消耗导致大气中CO2浓度不断增加,加剧了全球气候和环境的恶化。然而,CO2作为一种可再生的碳资源,具有无毒、储量大、廉价、易得等优点,在合成化工领域具有巨大的应用潜力[1-3]。因此,各国科研人员积极开发CO2转化技术,利用其合成高附加值的低碳化学品和燃料,从而缓解能源危机和温室效应[4-6]。其中,电化学CO2还原(CO2RR)技术能够通过控制电子还原途径将CO2转化为CO、甲酸/甲酸盐、甲醇、CH4、C2H4等低碳化学品和燃料,被认为是具有环境和经济效益的新策略[7-10]。但是CO2还原的活化能很高,在常温常压下需要很高的过电势,同时反应速率缓慢、选择性较低。因此,开发新型催化剂和电化学稳定的电解液体系,来降低电催化CO2还原的过电势,提高还原反应的电流密度和还原产物的选择性成为近年来该领域研发的重点。目前常用的贵金属催化材料具有成本高昂、催化稳定性差、环境毒性大等缺点[11];同时常用的KHCO3水性电解液存在CO2溶解度小、电催化CO2还原的电流密度过小、还原产物法拉第效率低等问题,进而限制了其规模化的使用[12-13]。
近年来,无毒且非贵重的金属硫化物在电催化CO2还原的研究中受到越来越多的关注。其中二硫化钼(MoS2)是一种具有三明治层状结构的晶体,两个S原子层中间插入一个钼(Mo)原子层,层与层之间通过范德华力结合,具有优异的物理化学性能,在润滑、储能、催化等领域拥有非常广阔的应用潜力[14-18]。2014年ASADI et al[14]首次报道了一种层状堆积MoS2在1-乙基-3-甲基咪唑四氟硼酸盐([Emim]BF4)离子液体(即摩尔分数为4%[Emim]BF4和96%的水)中表现出优异的CO2还原性能,该研究采用形貌调控、过渡金属掺杂和复合N掺杂碳等方法调节MoS2边缘电子密度来增强CO2还原的催化性能和还原选择性,证明了Mo元素在层状边缘具有更高的催化活性。例如,ABBASI et al[15]使用化学气相沉积法合成了金属铌(Nb)掺杂的MoS2纳米阵列催化剂,其CO产物的转化频率(turnover frequency)比原始MoS2纳米阵列提高了一个数量级,比Ag纳米颗粒提高了两个数量级。LI et al[16]合成了一种ZIF-67衍生N掺杂碳(NC)和边缘暴露的2H-MoS2(NCMSH)多级中孔催化剂,MoS2大量暴露的边缘提供了大量活性中心,在-0.7 V vs. RHE过电位下电流密度达到-34.31 mA/cm2,CO法拉第效率达到92.68%.SUN et al[17]报道了不同元素的双金属硫化物(Mo-Bi,Mo-Ag,Mo-Cu)在0.5 mol/L的1-丁基-3-甲基咪唑四氟硼酸酯(([Bmim]BF4)-MeCN)电解液中电催化CO2还原的催化性能。结果发现,Mo-Bi双金属硫化物在电流密度为-12.1 mA/cm2时,CO2电化学还原为甲醇的法拉第效率可达到71.2%,远高于文献报道的电流密度。电极的优异性能归因于Mo和Bi在生产甲醇中的出色协同作用。Mo-Bi BMC/CP电极的高电催化选择性可以归因于Mo和Bi在合成甲醇中的协同作用。相比KHCO3水性电解液,离子液体作为新型介质和材料时,由于其酸碱可调、结构可设计、CO2溶解度高,因此在CO2转化反应尤其是电化学还原研究中展现出广阔的应用前景。侧链带氨基的咪唑类离子液体具有一定碱性,对CO2的吸收速率、溶解度相对较高,由离子液体与水构成的电解液体系有望助力提高反应动力学和选择性[19-24]。
本文基于Mo-Bi纳米双金属硫化物的高CO2催化还原活性,有望在其协同催化作用下表现出更好的催化活性和定向选择性调控能力,但考虑到材料自身导电性能较差,故本论文设计合成了碳纳米管(CNTs)原位负载Mo-Bi-S的Mo-Bi-S@CNTs复合双金属硫化物纳米材料颗粒,考察了水热温度对催化剂形貌结构的影响,在此基础上考察催化剂电极以及在[NH2-emim]BF4-H2O(ω=40%)电解液中CO2电催化还原制备甲醇的性能。
1 实验部分
1.1 实验试剂与仪器
实验试剂: 碳纳米管(CNTs,直径30~50 nm,长度0.5~2 μm,南京先丰纳米材料),1-胺乙基-3-甲基咪唑四氟硼酸盐(C6H12BF4N3,[NH2-emim]BF4,上海成捷化学有限公司),Nafion溶液(5%,美国杜邦公司)。其余试剂均为分析纯,购买于国药集团山西有限公司。
实验仪器:材料晶相结构采用X-射线粉末衍射仪(DX-2700型)进行表征,辐射源为Cu Kα(λ=0.154 056 nm),管电压40 kV,管电流100 mA,扫描范围2θ=5°~85°,扫描速度8(°)/min.材料的形貌采用扫描电子显微镜(Nano SEM-430)进行检测,放大倍数在10~200 000之间,分辨率为0.8 nm(15 kV),加速电压20~30 kV之间。采用透射电子显微镜(JEM-2010)对材料结构进行进一步观察,加速电压为200 kV.催化剂电催化性能采用多通道恒电位仪(美国PAR公司,VMP 3型)进行测试。电化学催化CO2还原产物采用气相色谱仪(上海海欣色谱仪有限公司,型号GC7290)进行定性定量分析。
1.2 实验方法
1.2.1CNTs的预处理
CNTs在浓硝酸溶液中150 ℃回流反应2 h,反应结束后,抽滤,用去离子水彻底洗涤,于80 ℃下干燥得到表面预处理的CNTs,留以备用。
1.2.2Mo-Bi-S@CNTs的制备
以上述表面预处理的CNTs为载体,四硫代钼酸铵((NH4)2MoS4)和五水硝酸铋(Bi(NO3)3·5H2O)为反应物,利用水热法制备Mo-Bi-S@CNTs复合型纳米双金属硫化物催化剂,其中Mo、Bi、C摩尔比为1∶1∶4。制备过程如下:将0.1 g(NH4)2MoS4与0.02 g表面预处理的CNTs加入40 mL去离子水中,充分搅拌混合30 min;在20 mL去离子水中溶解0.187 g Bi(NO3)3·5H2O;将以上两种溶液移至Ar除氧后的反应釜内并继续搅拌30 min得到反应混合液;混合液分别在185 ℃、200 ℃、215 ℃、230 ℃的水热条件下反应12 h,冷却后进行洗涤干燥制得Mo-Bi-S@CNTs.
1.2.3工作电极的制备
称取4 mg催化剂、0.95 mL乙醇、5 μL Nafion溶液,超声处理15 min,得到混合均匀的悬浊液,使用微量移液器取10 μL悬浊液涂覆于玻碳电极(Φ=6 mm)上,红外干燥制得薄膜工作电极。
1.2.4电化学性能测试
采用线性伏安测试(LSV)、计时电流测试(CA)在H型隔膜电解池中室温(251 ℃)条件下进行三电极电化学表征,阳极室电解液为0.1 mol/L硫酸溶液,阴极室电解液为[NH2-emim]BF4-H2O(ω=40%),工作电极为上述薄膜工作电极,饱和甘汞电极(SCE)为参比电极,铂电极(1 cm×1 cm)为对电极。实验时首先在阴极室离子液体电解液中通入Ar去除溶解氧,再通入CO2置换1 h使其达到饱和,在测试过程中CO2流速稳定为0.1 L/h.文中所给电位数据均相对于SCE参比电极。
1.2.5还原产物的检测
气、液相产物,采用气相色谱进行定性定量检测分析,用公式(1)计算产物的法拉第效率:
(1)
式中:η为法拉第效率;z为电还原CO2电子转移数;n为还原产物的物质的量,mol;F为法拉第常数,C/mol;I为反应电流,mA;t为反应时间,s.
2 结果与讨论
2.1 XRD分析
图1是不同水热温度制备得到的Mo-Bi-S@CNTs的XRD谱图。从图中可以看出,185 ℃制备得到的样品只在24.9°、28.6°、46.6°三个位置出现了Bi2S3(130)、(211)、(431)的特征衍射峰(JCPDS 17-0320).当水热温度提高至200 ℃以上时,分别在33.5°和35.8°出现了MoS2(101)和(102)的特征衍射峰(JCPDS 17-0744)。以上结果表明,在较低温下可反应制得低结晶度的Bi2S3,随着水热温度升高,Bi2S3和MoS2衍射峰强度更高、更尖锐,说明结晶度有所提高。这是由于水热温度上升,(NH4)2MoS4分解速率逐渐加快,MoS2和Bi2S3生成速率提高,晶化时间延长,结晶度相应提高[25]。另外,所有样品均在2θ=26.02°出现尖锐的CNTs的(002)衍射峰。根据谢乐公式计算Bi2S3在扫描角度2θ为28.06°时的晶粒尺寸(表1),由计算结果可知水热温度为200 ℃得到的样品晶粒较小,约为12 nm.

图1 不同水热温度Mo-Bi-S@CNTs样品的XRD谱图Fig.1 XRD patterns of Mo-Bi-S@CNTs at different hydrothermal temperatures

表1 不同样品的晶粒尺寸Table 1 Crystallite size of the catalysts
2.2 SEM、XPS与TEM分析
不同水热温度制备样品的微观形貌结构如图2所示。从图2(a)中可以看出,当水热温度为185 ℃时,Mo-Bi-S@CNTs展示出纳米纤维结构,直径约为50 nm,纤维的周围散落着一些小尺寸的颗粒。当水热温度为200 ℃时,样品呈现为棒状与片状颗粒的复合物,如图2(b)所示。215 ℃、230 ℃水热温度制备的样品,均呈现出较细的棒状结构纳米材料与大量无规则形状结构颗粒的复合物,如图2(c)和2(d)所示。据推测,随着水热温度的升高,棒状结构颗粒逐渐变细,然后融合成片状结构,尺度变小甚至粉化,与纳米颗粒结合形成纳米片状结构,纳米颗粒由分散态逐渐转变为部分纳米颗粒聚集的状态。

图2 不同水热温度Mo-Bi-S@CNTs样品的SEM照片Fig.2 SEM images of Mo-Bi-S@CNTs at different hydrothermal temperatures
图3为Mo-Bi-S@CNTs-200 ℃样品的XPS图谱。图中各峰分别对应Mo4+(Mo 3d3/2:235.5 eV;Mo 3d5/2:231.8 eV,228.6 eV),Bi3+(Bi 4f5/2:164.2 eV,159.4 eV;Bi 4f7/2:163.7 eV,158.3 eV),以及S(S 2s:225.6 eV;S 2p:161.3 eV,162.6 eV).测试结果证明该样品为MoS2和Bi2S3,同时Mo 3d和Bi 4f特征峰与标准峰相比均有一定程度的偏移,更进一步说明MoS2和Bi2S3具有强相互作用,不是单纯的混合状态。

(a) Mo 3d3/2;(b) Mo 3d5/2;(c) S 2s;(d) Bi 4f5/2;(e) Bi 4f7/2;(f) S 2p图3 Mo-Bi-S@CNTs-200 ℃的XPS图谱Fig.3 XPS spectra orbits of Mo-Bi-S@CNTs-200 ℃
根据XPS能谱表(表2)可知,所制备催化剂表面Mo元素与Bi元素的摩尔比约为1∶1,与投入物料比相一致,符合实验预期的结果。
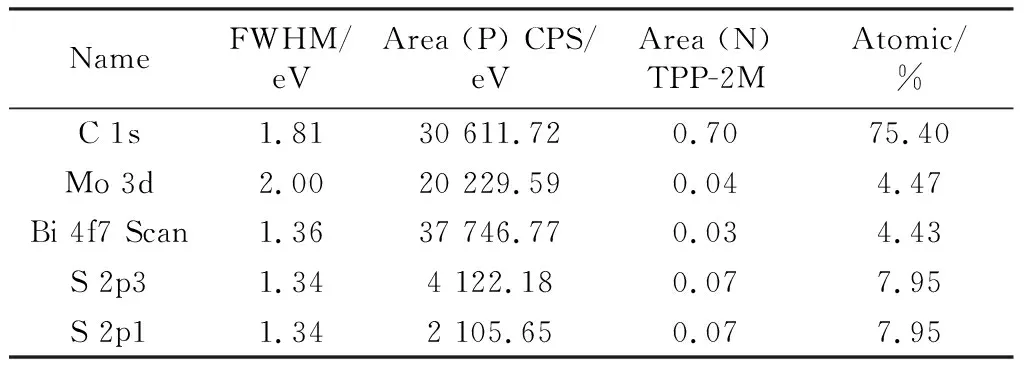
表2 XPS能谱表Table 2 XPS peaks table
采用透射电子显微镜进一步表征Mo-Bi-S@CNTs-200 ℃的微观结构。从图4(a)中可以看出,呈现出不规则形状颗粒的Mo-Bi-S聚集态包覆在CNTs的表面,使其直径增加到100 nm左右。进一步从图4(b)中可以看出,Mo-Bi-S颗粒粒径约在10~20 nm.二元金属硫化物Mo-Bi-S复合纳米颗粒成功负载到CNTs上。

图4 Mo-Bi-S@CNTs-200 ℃的TEM图Fig.4 TEM images of Mo-Bi-S@CNTs-200 ℃
2.3 电催化还原性能测试
图5是不同温度下制备的Mo-Bi-S@CNTs样品在Ar饱和和CO2饱和的[NH2-emim]BF4-H2O(ω=40%)电解液中的电位线性扫描伏安曲线。从图5(a)中可以看出,不同样品在Ar饱和条件下起始还原电位基本相同,约为-0.22 V,推测当电位下降到该电位以下时,电极上发生析氢反应。当扫描电位进一步负移,析氢电流逐渐增加。比较不同样品的还原电流密度可知,随着水热温度的提高,-0.50 V对应的析氢电流密度先增大后减小,其中,Mo-Bi-S@CNTs-200 ℃电极在电位为-0.50 V vs.SCE时,析氢电流密度为-0.26 mA/cm2.图5(b)为不同样品在CO2饱和的[NH2-emim]BF4-H2O(ω=40%)电解液中的线性伏安曲线。从图中可以看出,不同样品的起始还原电位基本相同,约为-0.34 V vs.SCE,比无CO2电解液中的电位负移0.12 V.当扫描电位进一步负移,电极还原电流从-0.4 V vs.SCE开始迅速增加,其增加的幅度远高于无CO2电解液中的情况。同时比较不同样品在相同电位下的还原电流密度可知,Mo-Bi-S@CNTs材料制备的电极随着制备水热温度的提高,样品的还原析氢电流先增大后减小。其中,Mo-Bi-S@CNTs-200 ℃电极的电流密度最高,在电位为-0.50 V vs.SCE时,析氢电流密度达到-11.28 mA/cm2.
Ar饱和(a)和CO2饱和(b)的[NH2-emim]BF4-H2O(ω=40%)电解液,图(c)为图(b)对应的Tafel曲线图5 不同水热温度制备的Mo-Bi-S@CNTs的伏安曲线Fig.5 LSV curves of Mo-Bi-S@CNTs
根据图5(b)绘制Tafel曲线(图5(c)),由Tafel公式(公式(2))可计算出对应的斜率b和交换电流密度i0,如表3所示。
η=i0+blog|i| .
(2)
式中:η为该电极反应的过电位,V;|b|的大小反映了电极极化过程中的受阻情况,mV·decade-1,对于析氢反应,该数值越小表示电极在极化过程中受到的阻力越小;i0反应了在电极反应中所需要的外电流密度,mA·cm-2,该数值越大表示该电极反应所需要的外电流密度越小,即该电极反应所需的推动力越小。

表3 Tafel斜率b和交换电流密度i0Table 3 Tafel slope and exchange current density
由以上结果可知,200 ℃水热样品的Tafel斜率b相对较小,交换电流密度i0相对较大,该样品在本反应中的电化学动力学较优,CO2电催化反应阻力较低。
进一步对Mo-Bi-S@CNTs电极在[NH2-emim]BF4-H2O(ω=40%)电解液中的CO2还原产物进行分析测定发现,催化剂材料的制备温度对产物的法拉第效率影响较为显著。
图6是Mo-Bi-S@CNTs电极电催化CO2还原产物(CO、CH3OH和CH4)的法拉第效率图。随着还原电位的增加,产物CO的法拉第效率缓慢降低,其中Mo-Bi-S@CNTs-200 ℃电极在低于-0.3 V vs.SCE的电位后,没有检测出产物CO,如图6(a)所示。产物CH3OH的法拉第效率随着还原电位的提高先增大后减小,其中200 ℃制备的催化剂电极的CH3OH法拉第效率高达63%,而Mo-Bi-S@CNTs-185 ℃电极没有检测出产物,如图6(b)所示。产物CH4只在高还原电位下生成,只有Mo-Bi-S@CNTs-200 ℃电极能够生成CH4,如图6(c)所示。综上所述,Mo-Bi-S@CNTs-200 ℃电极在电解液[NH2-emim]BF4-H2O(ω=40%)中电位为-0.3 V vs.SCE条件下主产物CH3OH的法拉第效率高达63.0%.然而Mo-Bi-S@CNTs-185 ℃样品制备电极的产物只有CO.

图6 不同水热温度制备的催化剂电催化CO2还原产物法拉第效率图Fig.6 Faradaic efficiencies of CO2 reduction products
由于MoS2界面边缘适于吸附态H(Hads)的生成,Bi3+间的界面可以得到较稳定的吸附态CO(COads),因而在Mo-Bi-S@CNTs复合催化剂中Hads由Mo活性位承担生成,COads由Bi活性位承担生成,与[NH2-emim]+协同作用于CO2还原,Hads与COads在电极表面进一步加氢进行结合形成CHOads,继续氢化得到CH3OH,其反应途径推断为CO2→CO2·→COads→CHOads→CH3Oads→CH3OH.在此过程中,氢源来自于电解液中的水,因此析氢反应作为竞争反应始终存在,并对甲醇的生成有一定的影响[14-16]。
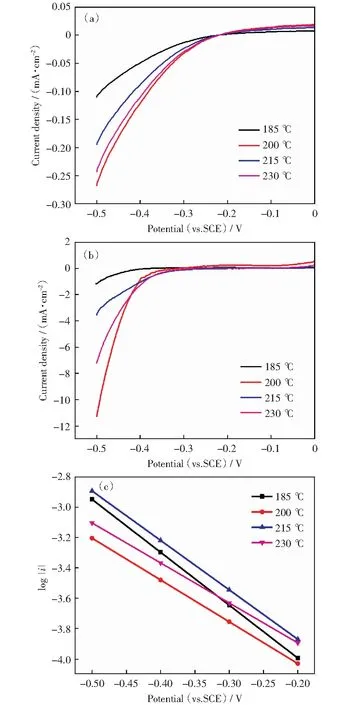
图7是不同温度制备的Mo-Bi-S@CNTs催化剂在[NH2-emim]BF4-H2O(ω=40%)电解液中电位为-0.3 V vs.SCE下的计时电流曲线。从图中可以看出,在该电位下,CO2还原电流密度迅速降低,在0.5 h后基本保持不变。同时随着制备温度的增加,还原电流先增大后减小。Mo-Bi-S@CNTs-200 ℃电极在电解池连续运行6 h后电流密度仍然保持在-13.5 mA/cm2左右。这些结果表明Mo-Bi-S@CNTs-200 ℃催化剂具有良好的稳定性。
图7 Mo-Bi-S@CNTs在[NH2-emim]BF4-H2O(ω=40%)电解液中-0.3 V vs.SCE电位下的计时电流曲线Fig.7 Chronoamperometry curves of Mo-Bi-S@CNTs in [NH2-emim]BF4-H2O(ω=40%) electrolyte at -0.3 V vs.SCE
3 结论
1) 采用水热法在不同水热温度下制备得到了碳纳米管(CNTs)原位负载Mo-Bi-S双金属硫化物的纳米颗粒。当水热温度为200 ℃时,制备的Mo-Bi-S@CNTs催化剂的钼铋元素摩尔比为0.92∶1,颗粒尺寸较小,约为10~15 nm,并且均匀地负载在CNTs上。温度对复合催化剂的结晶形态、化学组成和颗粒大小有显著的影响。
2) 研究催化剂电极在[NH2-emim]BF4-H2O(ω=40%)电解液中的电催化还原CO2制备甲醇的性能。在反应电位为-0.3 V vs. SCE时,作为主产物甲醇的法拉第转化效率高达63.0%;且电解池连续运行6 h后电流密度保持在-13.5 mA/cm2,表明Mo-Bi-S@CNTs-200 ℃在电催化CO2制备甲醇反应中具有优秀的催化活性、甲醇选择性以及稳定性。
