基于系统生物学的非小细胞肺癌气阴两虚证多组学联合分析研究
2021-11-25傅晓璇马冠君张爱琴
陈 卓 钱 祥 傅晓璇 马冠君 张爱琴
证候的生物学基础研究是中医现代化研究的关键部分,也是阐明中医科学本质的核心环节。在中医传统理论指导下,利用多组学技术可以从不同的层面探讨相互作用及调控关系[1]。“组学”强调从整体角度研究生物体的功能,这与中医“整体观”高度一致。综合各组学技术进行中医证候研究以获得该证候的基因表达谱、蛋白质图谱与代谢群谱,可能会更全面、更有效地揭示证候的本质与规律[2]。肺癌是目前世界范围内肿瘤发病率和死亡率最高的恶性肿瘤[3],气阴两虚证是其常见证候。因此,本研究运用16S rDNA和液相色谱-质谱联用技术(LC-MS 技术)对非小细胞肺癌(NSCLC)气阴两虚证进行蛋白质组学、代谢组学和肠道微生态学的研究,旨在为NSCLC 中医证候的诊断提供生物学基础,为今后开展中医药抗癌治疗提供实验依据。
1 资料与方法
1.1 一般资料 本次研究对象来自2019 年3 月—2019 年9 月浙江省肿瘤医院胸部外科门诊就诊患者13例,拟行肺癌根治术(入组前未做任何治疗),由中医科两名副主任以上中医师同时辨证为气阴两虚证,其中男3例,女15例,年龄46~75(59.23±8.67)岁,TNM 分期[4](恶性肿瘤国际临床病期分类):原位癌1例,Ⅰa 期9例,Ⅰb 期1例,Ⅱb 期1例,Ⅲa 期1例。同时期招募健康志愿者15 名,要求经普通体检及实验室检查排除心脑血管、肝肾、内分泌等重大疾病且近3 周内无感染性疾病、无抗生素治疗史。本临床研究通过浙江省肿瘤医院医学伦理委员会审核(批准号:IRB-2018-219)。
1.2 诊断标准 经组织病理学和/或细胞学证实为NSCLC[5]。符合《中医临床诊疗术语·证候部分》[6]气阴两虚证诊断标准:神疲乏力,口干少饮,舌质红或淡,脉细弱等为常见症的证候。
1.3 纳入、排除及脱落标准 纳入标准:(1)符合诊断标准;(2)年龄18~70 岁;(3)无其他恶性肿瘤个人史;(4)患者本人同意并签署知情同意书。排除标准:(1)近3 周内使用过抗生素治疗;(2)有精神疾病,无完全民事行为能力。脱落标准:(1)患者依从性差;(2)患者要求终止试验。
1.4 观察指标及方法
1.4.1 肠道微生态分析 称取粪便样本200mg,加入30mL 缓冲溶液,充分振荡混匀,1000r/min 离心5min 后收集沉淀,以10mL 缓冲液重悬。使用QIAamp Fast DNA Stool Mini Kit(QLAGEN-51604)对粪便微生物DNA 进行提取。待提取后,用琼脂糖凝胶电泳和Thermo NanoDrop 2000 紫外微量分光光度计进行总DNA 质检。将样本进行16S V3-V4区域扩增,引物信息为:上游引物序列为:341F(5'-CCTACGGGNGGCWGCAG-3';下游引物:805R(5'-GACTACHVGGGTATCTAATCC-3'(杭州晶佰生物技术有限公司提供)。将稀释后的基因组DNA 作为模板,使用Phanta Max Master Mix(Vazyme-P525)高保真酶进行聚合酶链式反应(PCR)。PCR 的反应条件是:95℃预变性3min;95℃变性30s,55℃退火30s,72℃延伸45s,重复25 个循环;最后72℃持续5min。PCR 反 应 体 系 为:2 ×Phanta Max Master Mix(Vazyme)25μL,引物(10μm)各为2μL,加入ddH2O至50uL。取PCR 产物1.5μL,使用2%琼脂糖胶,置于120V 电压下持续电泳20min 后使用凝胶成像系统进行紫外成像拍照。
文库质检合格后,使用量子位(Qubit)对混合文库pool 进行浓度定量,并依据每个样品的数据量要求,进行相应的比例混合,最后运行测序仪(Miseq)测序程序。将序列完全一样的原始测序数据(Clean Reads)根据其丰度大小进行排序,将其中的Singletons 过滤掉,利用Usearch 在0.97 相似度下进行聚类,对聚类后的序列进行嵌合体过滤,得到用于物种分类的奥图(OTU),最后将所有Clean Reads 比对到OTU 序列上,将能比对上OTU 的Reads 提取出来,得到最终的比对后数据(Mapped Reads)。
1.4.2 蛋白组学分析 使用抗凝真空采血管收集空腹外周血5mL,静置20min 后采用3000r/min 离心分离血清。在待测血清样本中加入裂解液,沸水中水浴15min。使用-20℃低温高速离心机14000r/min 离心15min 取上清液。分装样品,-20℃低温保存。鉴定每个样本中蛋白,进行电泳并定量各蛋白的浓度及总量。取蛋白提取物30μg,加入200μL 的8M 尿素充分混合,在20℃、14000r 离心15min。浓缩液用200μL 的8M 尿素在pH 8.5 的0.1 三羟甲基氨基甲烷盐酸盐(MTris-HCl)中稀释后进行离心。沉淀用100μL 的8M 尿素在pH 8.5 的0.1MTris-HCl 中稀释后再次离心。浓缩液经胰蛋白酶(酶蛋白比为1∶100,50mMABC)在37℃湿室消化12h 后离心并收集消化液,用50μL 的0.5M 氯化钠冲洗后再离心。将得到的溶液与10%的三氟乙酸结合并酸化。酸化后用C18TIPS(皮尔斯,Thermo Science entific)脱盐。根据制造商的说明进行活化、平衡、肽洗和洗脱。TIPS 用50%乙腈2 次漂洗活化,然后用0.1%三氟乙酸水溶液两次漂洗平衡。将富含多肽的溶液缓慢抽吸并由TIP 分配10 个循环。用0.1%三氟乙酸进行两次洗涤脱盐。脱盐肽用50μL 50%CAN 洗脱,洗脱产物真空干燥,再悬浮于30μL 的0.1%甲酸(FA)中。含脱盐肽(3μL)的溶液通过EASY-纳米LC 系统进行在线纳米流动液相色谱分离。将LC 连接到内径为100μm的2cm 预柱上,填充5μm C18 树脂。柱前反应后,用3μm C18 树脂填充75μm×15cm 毛细管柱。通过逐步梯度洗脱从柱前和分析柱上洗脱多肽。根据Maxquant(1.6 版)中的Uniprto 人类蛋白序列数据库搜索所有的原始文件。用Perseus 统计各亚组的折叠变化。数据框中大于2 倍变化(>2 or<0.5)的蛋白质组被标记为显著改变的蛋白质。
1.4.3 代谢组学分析 使用抗凝真空采血管收集空腹外周血5mL,静置30min 后采用3000r/min 离心分离血清,-80℃度低温冰箱保存。取血清样本200μL,在室温下冰上解冻,加入预先冷藏-20℃过夜的甲醇/乙睛(1∶1)600μL,涡旋30s,14000r/min 4℃离心15min,取上清液置离心管中,真空离心浓缩仪干燥。样本干燥后加入乙腈/甲醇(80/20)-水混合溶液(1∶1)复溶,涡旋60s,14000r/min 4℃离心15min,取上清液4μL 进UPLC-Q/TOF-MS 分析。
待测样品采用ExionLCAD 超高效液相色谱系统(UHPLC)ACQUITY UPLC HSS T3(100mm×2.1mm,1.8μm)色谱柱进行分离;进样盘温度:4℃;柱温:40℃;进样量:4μL;流动相组成A:乙腈(含0.1%甲酸),B:水(含0.1%甲酸);以0.3mL/min 的流速进行洗脱,洗脱梯度:0~1min,95%B;1~20min,95%~1%B;20~23min,1%B。样品经UHPLC 分离后用X-500R质谱仪(AB SCIEX)进行质谱分析。通过单、多维统计分析筛选得到具有显著性差异的离子(P<0.05),差异倍数(FC)>1.5 且VIP>1.0,作为潜在生物标记物。
1.4.4 肠道微生态-代谢组学联合分析 本项目通过对代谢组学筛选得到的差异二级代谢物和16SrDNA 测序分析获得的显著性差异属水平菌群进行Spearman 相关性分析,获得差异菌群与差异菌群,差异代谢物与差异代谢物,差异代谢物与差异菌群之间的关系。基于计算结果选择合适的筛选条件,获得最终的差异代谢物与差异代谢物的相关关系及网络图等。
1.4.5 蛋白组学-代谢组学联合分析 通过京都基因与基因组百科全书(KEGG)通路将代谢组学和蛋白质组学数据联合起来,找到参与同一生物进程(KEGG Pathway)中发生显著性变化的蛋白质和代谢物,快速锁定关键基因。主要分为以下几步:(1)蛋白质组与代谢组通过KEGG 代谢通路进行关联;(2)显著差异数据和调控关系对数据筛选;(3)基因本体论(GO)和KEGG 富集分析;(4)关联数据表达模式聚类;(5)关联数据网络图。
1.5 统计学方法 通过秩和检验找出不同组间有明显差异的肠道菌群,显著性筛选的阈值为P<0.05,同时对所有的P 值进行错误发现率(fdr)校验。在差异蛋白分析中,生物学重复组间比较,符合差异倍数为1.2倍(上下调)且P<0.05 筛选标准的蛋白质视为差异表达蛋白质,T-test 检验方式限于有生物学重复的样本两组之间的比较。在进行两组样本间的差异代谢物分析时,利用单变量分析可以直观地显示两样本间代谢物变化的显著性,从而帮助我们筛选潜在的标志代谢物(通常以FC>1.5 且P<0.05 作为筛选标准)。联合分析采用斯皮尔曼相关系数。
2 结果
2.1 微生态组间差异
2.1.1 维恩图 16SrDNA 测序分析结果显示,本研究中28 个样品共产生242 个OTU。两组样本有133个共有OTU,患者组有22 个独有的OTU,健康对照组有87 个独有的OTU。见图1。

图1 OTU 韦恩图
2.1.2 差异物种分析 组间群落差异分析(LDA EffectSize,LEfSe),LEfSe 采用线性判别分析(LDA)来估算每个组分(物种)丰度对差异效果影响的大小,找出对样品划分产生显著性差异影响的群落或物种。下图统计两组中有显著作用的微生物类群通过LDA(线性回归分析)后得到的LDA 分值。通过差异物种分析,两组之间有显著差异(P<0.05)的菌属,分别是罗斯氏菌、吉米菌、嗜沫凝聚杆菌、Fusicatenibacter、Intestinimonas、拉乌尔菌、克雷伯氏菌、养分缺乏菌、气球菌、肠杆菌,前5 种菌群在患者组的丰度水平明显高于健康对照组,后5 种菌群在患者组的丰度水平明显低于健康对照组。见图2。

图2 差异物种LEfSe 分析图
2.1.3 差异物种Spearman 相关系数分析 应用LEfSe 在各水平上或秩和检验在属水平上(或某一特定水平),选择丰度Top30 的差异物种,通过R 软件的corrplot 包绘制物种之间Spearman 相关性热图,并通过该热图可以发现物种之间重要的模式与关系。通过差异物种Spearman 相关系数分析发现肠杆菌(Enterobacteriaceae)与埃希氏菌属/志贺杆菌属(Escherichia/Shigella)、克雷伯氏菌存在正相关,链球菌属与放线菌属存在正相关,普氏菌属与柯林斯菌属存在负相关。见图3。
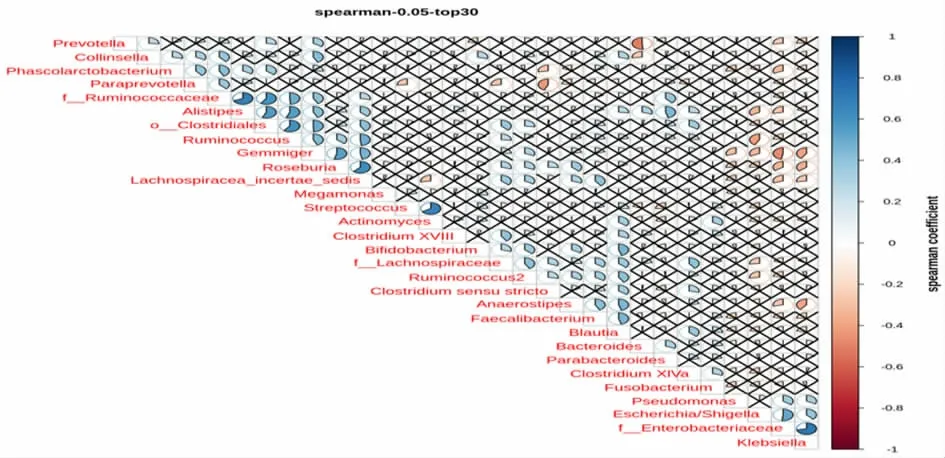
图3 丰度Top30 的差异物种相关性系数分析
2.2 蛋白组学组间差异
2.2.1 差异蛋白上下调统计分析 差异蛋白上下调频数统计用于判断不同实验条件下差异蛋白的个数。其中横坐标表示比较组信息,纵坐标表示上下调蛋白的数目,红色代表上调蛋白,蓝色代表下调蛋白,其中数字代表上下调蛋白的数目。蛋白定量分析结果发现,与健康对照组相比,患者组有15 个上调蛋白和5 个下调蛋白,共计20 个差异蛋白。见图4。

图4 差异蛋白图
2.2.2 差异蛋白基因功能分析 GO 的基本单位是term(词条、节点),每个term 都对应一个属性。GO 功能显著性富集分析首先把所有显著性差异表达基因向Gene Ontology 数据库的各term 映射,计算每个term 的基因数目,然后应用超几何检验,找出与整个基因组背景相比,在显著性差异表达基因中显著富集的GO 条目。见图5。

图5 GO 富集性散点图
2.2.3 差异蛋白KEGG 通路分析 通路显著性富集分析以KEGG 通路为单位,应用超几何检验,找出与整个基因组背景相比,在显著性差异表达基因中显著性富集的通路。见图6。
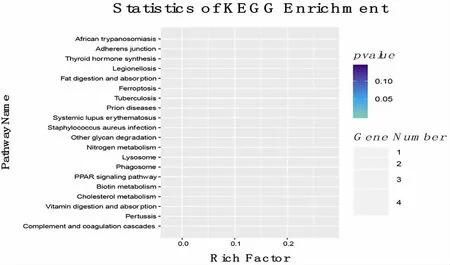
图6 KEGG 富集性散点图
2.3 代谢组学组间差异 本实验以单变量统计分析P<0.05和FC>1.5 的代谢物,作为具有显著性差异的代谢物,采用MetDNA和OSI/SMMS 软件进行代谢物结构鉴定,具体鉴定结果(见表1),差异代谢物强度对比(见图7)。本项目共鉴定出差异代谢物11 个,其中负离子模式下2 个,正离子模式下9 个。将得到的差异代谢物提交到Metaboanalyst 4.0 网站,进行代谢通路分析。结果显示,差异代谢物主要涉及亚油酸代谢、不饱和脂肪酸的生物合成、α-亚麻酸代谢、卟啉和叶绿素代谢、花生四烯酸代谢和色氨酸代谢等代谢通路,见图8。
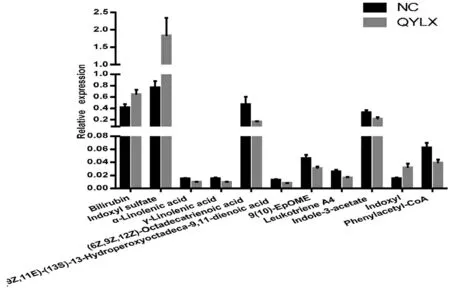
图7 差异代谢物强度对比
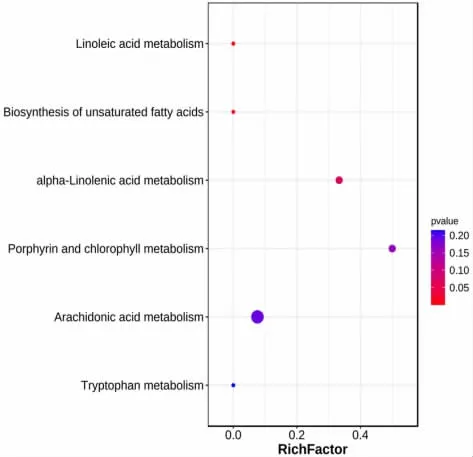
图8 差异代谢物富集代谢通路示意图
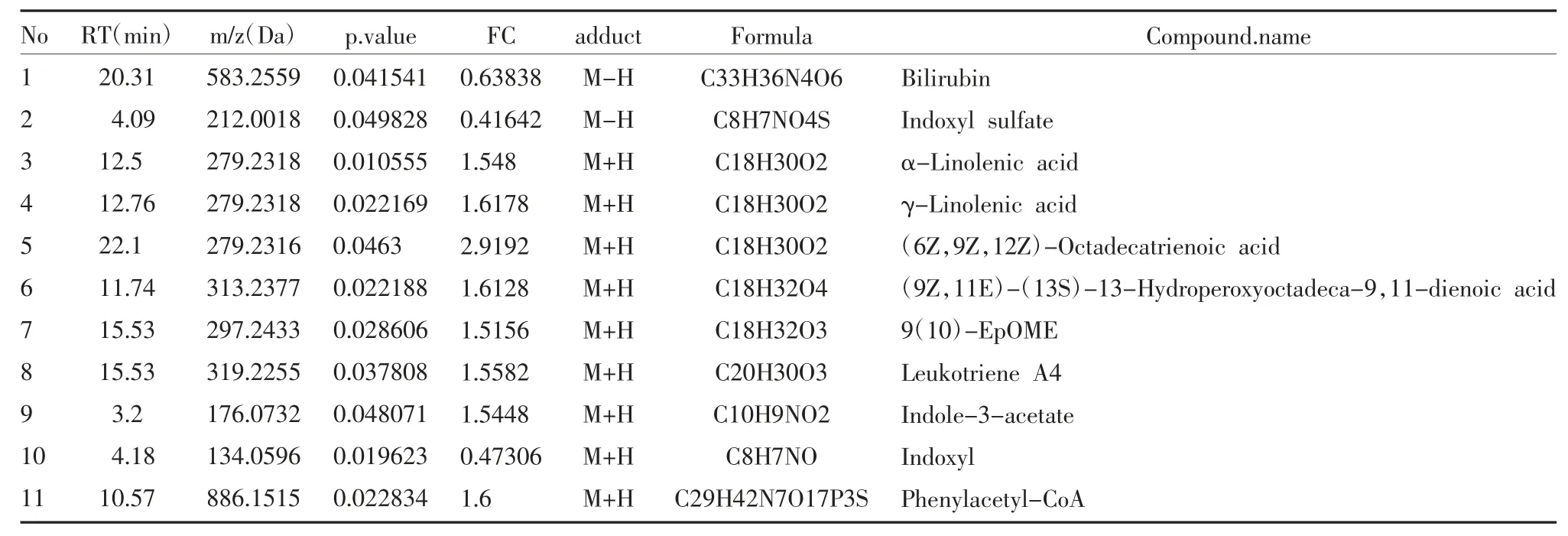
表1 差异代谢物列表
2.4 肠道微生态-代谢组学联合分析
2.4.1 差异代谢物-差异菌群相关性分析 通过菌群-代谢物联合分析,我们发现硫酸吲哚氧基与艾克曼菌、梭状菌存在正相关,与放线菌存在负相关。胆红素与克雷伯菌存在负相关,与酸氨基球菌存在正相关。见图9。
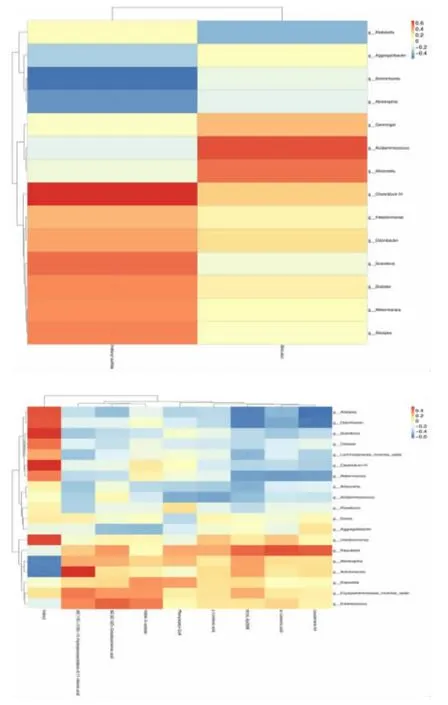
图9 差异代谢物-差异菌群关联热图
2.4.2 差异代谢物与菌群网络调控分析 基质金属蛋白酶-9(matrix metalloproteinase-9,MMP9)、纤连蛋白(fibronectin,FN1)、胰岛素样生长因子Ⅱ受体(cation-independent mannose-6-phosphate receptor,IGF2)、巨噬细胞集落刺激因子1 受体(macrophage colony-stimulating factor 1 receptor,CSF1R)、基质金属蛋白酶-9(matrix metalloproteinase-2,MMP2)、巨噬细胞集落刺激因子受体(platelet-derived growth factor receptor beta,PDGFRB)。见图10。

图10 差异代谢物与菌群网络调控图
2.5 蛋白组学-代谢组学联合分析 通过蛋白组学-代谢物联合分析,患者组差异蛋白基质金属蛋白酶-9(Matrix metalloproteinase-9,MMP9)、纤连蛋白(Fibronectin,FN1)、胰岛素样生长因子Ⅱ受体(Cation-independent mannose-6-phosphate receptor,IGF2)、巨噬细胞集落刺激因子1 受体(Macrophage colony-stimulating factor 1 receptor,CSF1R)、基质金属蛋白酶-9(Matrix metalloproteinase-2,MMP2)、巨噬细胞集落刺激因子受体(Platelet-derived growth factor receptor beta,PDGFRB)及下游代谢物雄烯二酮与癌症通路具有相关性。见图11。

图11 差异蛋白质与差异代谢物关联网路图
3 讨论
“组学”是系统生物学的重要组成部分,将不同层面的信息进行科学整合得到关联分析结果,有助于扩大视窗角度。气阴两虚证是肺癌的常见证候,临床主要表现为神疲乏力,口干少饮,舌质红或淡,脉细弱等症状[5]。肠道微生物是宿主不可忽视的一部分[7],中医药与肠道菌群的现代研究拓展了“脏腑学说”中“肺与大肠相表里”的理论[8]。在本研究中,相较于健康对照人群的肠道微生态,患者组存在明显差异,患者组有22 个独有的OTU,健康对照组有87 个独有的OUT。通过差异物种分析发现,两组间差异菌属共有10 种,其中罗斯氏菌、吉米菌、嗜沫凝聚杆菌、Fusicatenibacter、Intestinimonas 在患者组的丰度水平明显高于对照组,拉乌尔菌、克雷伯氏菌、养分缺乏菌、气球菌、肠杆菌在患者组的丰度水平明显低于对照组(P<0.05)。Intestinimonas 菌种属于瘤胃菌科,研究发现瘤胃球菌属在NSCLC患者肠道微生态中含量较健康人群高[9],这与本研究结果一致,但其与肿瘤发生发展的关系有待进一步探索。本研究组进一步通过差异物种Spearman 相关系数分析,发现肠杆菌(Enterobacteriaceae)与埃希氏菌属/志贺杆菌属(Escherichia/Shigella)、克雷伯氏菌存在正相关。本研究推测肺癌的发生发展可能与肠道中某些菌群的增加或者减少有关。我们假设可通过观察NSCLC患者气阴两虚型在肠道微生态方面的差异性,并研究NSCLC患者气阴两虚型的生物学基础,以此来阐释证候的科学内涵。
本研究采用LC/MS 技术代谢组学分析,通过两组比较共鉴定出差异代谢物11 个,其中负离子模式下2 个,正离子模式下9 个。差异代谢物主要涉及亚油酸代谢、不饱和脂肪酸的生物合成、α-亚麻酸代谢、卟啉和叶绿素代谢、花生四烯酸代谢和色氨酸代谢等代谢通路。其中色氨酸代谢通过增加肿瘤细胞的恶性增殖和抑制抗肿瘤免疫反应来促进肿瘤进展,在肿瘤的发生进展中扮演重要角色[10]。
与健康对照组比较,患者组有15 个上调蛋白和5 个下调蛋白,共计20 个差异蛋白。通过基因功能分析及KEGG 功能分析,与肺癌相关的差异蛋白主要涉及在免疫反应的调节和癌症转录失调通路。其中CSF1R 是肿瘤相关巨噬细胞中的一个重要免疫信号通路。CSF1R 在正常组织中可协助巨噬细胞抑制受损组织处的免疫反应,同时促进血管再生来加速愈合,但是在癌症中这个功能却被用来促进肿瘤生长。此外,在小鼠模型中,发现CSF1R 抑制剂能够通过对肿瘤相关巨噬细胞进行重新编程,提高抗原呈现和促进抗肿瘤T细胞反应[11]。MMPs 作为目前已知能降解细胞外基质的重要酶类,在介导肿瘤血管新生、转移和侵袭等过程中发挥重要作用。MMP-2/9 更被认为是肿瘤发生发展过程中潜在的生物标志物,研究表明MMP-2/9 的表达与NSCLC、直肠癌、神经母细胞瘤、甲状腺乳头状癌等多种疾病进展相关[12]。本研究进一步通过蛋白组学-代谢组学联合分析发现,其中6 个差异蛋白及代谢产物雄烯二酮富集在癌症的相关通路。基础研究表明,雄烯二酮对肺癌细胞、前列腺癌细胞、乳腺癌细胞等具有一定的抑制作用[13]。
多组学研究是开启中医证候研究之门的钥匙,本研究在中医传统理论的指导下,通过多组学联合分析发现气阴两虚型NSCLC 具有特异的肠道菌群、蛋白质和代谢产物,且与肿瘤的发生进展密切相关。这有助于帮助我们阐明气阴两虚证的科学本质,同时对肺癌机理研究、确定致病靶点起到推动作用,为中医证候研究提供新思路。
