高效液相色谱法测定罗城“桂葡1 号”毛葡萄酿酒过程中9 种酚类物质的变化
2021-11-23郝俊光梁振荣林敏清龙玉凤梁城杰
郝俊光,梁振荣,银 书,林敏清,龙玉凤,唐 越,梁城杰,陈 静,*
(1.北部湾大学食品工程学院,钦州市食品风味分析与调控重点实验室,广西钦州 535011;2.广西天龙泉酒业有限公司,广西河池 546400)
毛葡萄(Vitis quinnquangularisRehd)是中国三大野生葡萄资源之一[1],其根系发达,抗旱、耐贫瘠、耐湿热高温、抗病性强,适合在石漠化的山区种植,可以帮助南方山区农民增收致富[2]。毛葡萄浆果富含氨基酸、微量元素、多种维生素、多酚类物质和SOD 抗衰老元素等,抗氧化活性较高[3],适合酿制葡萄酒[4]。国内各省利用当地毛葡萄野生种质资源进行了种质创新,培育出了许多酿酒葡萄新品种,例如两性花毛葡萄改良新品种“NW196 ”、“野酿2 号”、“桂葡1 号”、“桂葡2 号”、“云葡1 号”等,为毛葡萄酒的优质化和多样性酿造奠定了基础[4]。
酚类物质组成复杂多样,主要包括类黄酮类物质和非类黄酮类物质。其中类黄酮类物质又分为花色苷、黄烷醇类、黄酮醇类以及它们的衍生物;非类黄酮类物质又分为酚酸、芪类物质以及它们的衍生物[5]。类黄酮化合物是一类天然抗氧化物,具有抗氧化、抗炎症、抗癌、软化血管等保健功能,对葡萄酒的色泽、感官品质也起重要作用[6−8]。葡萄酒中酚酸类化合物含量丰富,具有抗氧化、抗炎、抑菌、抗糖尿病、抗癌等生物活性[8−10]。白藜芦醇作为葡萄的主要芪类物质,是一种天然的植物抗毒剂,具有抗肿瘤、防心血管疾病、消炎、杀菌、防衰老等功能[8,11]。花色苷是葡萄酒的主要呈色物质,而其它酚类物质起不同的辅色作用[8,12]。葡萄中的酚类物质主要分布在果皮中,其次是种子和果肉中[13−14]。葡萄酒的酚类物质虽然源自葡萄,但具体组成成分和含量受到品种[15−16]、农事[17−18]、酿造工艺[19−21]、陈化工艺[22]等的影响。目前,酚类物质的检测手段较多,而反相液相色谱一直是检测主要酚类物质的首选[5,23−25]。随着高精度质谱的应用,葡萄酒中越来越多的新酚类物质被认识[8,25−26]。
罗城毛葡萄2016 年获国家农产品地理标志认证[27],而“桂葡1 号”是广西农科院通过欧美酿酒葡萄和罗城野生毛葡萄杂交得到的适合南方气候种植的优质两性花酿酒葡萄[28],已在当地推广种植上万亩。深入研究毛葡萄酿酒过程中主要酚类物质的组成和变化,对生产优质毛葡萄酒产品十分重要。目前关于毛葡萄酿酒工艺的报道较多[4,28−29],但对毛葡萄酿酒过程酚类物质的变化研究较少[28]。本研究拟采用Waters Atlantis® T3 反相色谱柱开发包括芦丁、反式白藜芦醇等9 种代表性酚类物质的检测方法,进而对两个发酵温度下“桂葡1 号”毛葡萄酿酒过程中酚类物质的变化进行跟踪,以期为“桂葡1 号”毛葡萄酿酒提供数据支持。
1 材料与方法
1.1 材料与仪器
“桂葡1 号”毛葡萄 广西大益生态酒业有限公司提供,糖度11.5 °Brix、色泽紫黑、籽粒饱满、无霉烂损伤;焦亚硫酸钾、白砂糖、通用果酒酵母 食品级,烟台帝伯仕有限公司;UT 高温型复合果胶酶食品级,诺维信(中国)生物技术有限公司;矢车菊素-3-O-葡萄糖苷、绿原酸、阿魏酸、虎杖苷、芦丁、水杨酸、白藜芦醇、槲皮素、山奈酚、乙腈、乙酸 均为色谱纯,上海麦克林生化科技公司。
Waters Atlantis® T3 色谱柱(4.6 mm×250 mm,5 μm)、Waters Alliance 2695HPLC 分离单元、2996 PDA 检测器、Empower 工作站 美国Waters 仪器公司;DK-98-II 电热恒温水浴锅 天津市泰斯特仪器有限公司;Eppendorf INNOVA43R 落地式低温摇床 美国New Brunswick 生命科学仪器及设备公司;H1850 高速离心机 湖南湘仪实验室仪器开发有限公司;ME204E 电子天平 梅特勒-托利多仪器(上海)有限公司;Cascada I 实验室超纯水系统 美国PALL 公司;0.22 μm SLGP 033RB 针头滤膜 美国Millipore 公司;5 L 不锈钢发酵罐 烟台帝伯仕有限公司。
1.2 实验方法
1.2.1 毛葡萄酒的发酵 工艺流程[21,28]:除梗→洗净→称量→护色挤汁→酶解→活化酵母、接种→调糖→装罐→开始控温发酵→每天“压帽”三次→3 d 后倒罐→发酵15 d
操作要点为:
护色挤汁:往洗净的3 kg 毛葡萄中加入0.6 g焦亚硫酸钾,使焦亚硫酸钾达到200 mg/L,破碎挤汁,装入带取样龙头的5 L 不锈钢发酵罐中。
酶解:在毛葡萄浆中加入300 μL 复合果胶酶(UT),然后在50 ℃的水浴锅中酶解2 h。
活化酵母与接种:将0.6 g 通用果酒酵母加入100 mL 5%的糖水,然后在30 ℃水浴锅中活化30 min,接种到葡萄浆中。
调糖:在接种的葡萄浆中加入白沙糖320 g 使糖度调至 22 °Brix,封盖,并在发酵栓中加水。
发酵:将不锈钢发酵罐置于恒温培养箱中,分别在22、28 ℃下发酵,发酵第3 d 通过取样龙头进行皮渣分离并倒罐至新的发酵罐中。每个温度进行两个发酵平行实验。
1.2.2 定性标准溶液的配制 称取适量物质,用去离子水配制成矢车菊素-3-O-葡萄糖苷48 mg/L、绿原酸8 mg/L、阿魏酸8 mg/L、虎杖苷8 mg/L、芦丁80 mg/L、水杨酸40 mg/L、反式白藜芦醇8 mg/L、槲皮素8 mg/L、山奈酚8 mg/L 的单标,用于不同酚类物质保留时间和峰形的确定。将上述单标溶液等体积混合后用于色谱条件的优化,在不同色谱条件下色谱峰的辨识基于单标所获取的峰形、强度和出峰顺序。
1.2.3 色谱的基本操作条件 色谱柱:Waters Atlantis®T3 色谱柱(4.6 mm×250 mm,5 μm);流动相A:乙腈溶液;流动相B:1%乙酸溶液;洗脱方式:线性梯度;流速:1 mL/min;柱温:30 ℃;进样量:10 μL;检测波长:280、306、320、360 nm。
1.2.4 色谱条件的优化和酚类物质的定性 色谱条件的优化包括各物质的定量波长的确定和梯度洗脱条件(见表1)的优化。首先在梯度二条件下进行各物质在270~400 nm 波长下的紫外二极管阵列扫描,选取适宜的较大吸收波长作为该物质的检测波长。然后,对混合定性标准样品在选定的检测波长下进行分离优化,共进行了10 余次调整,仅以表1 中列出的3 个洗脱梯度说明优化效果。将各目标物完全分离、检测周期短的洗脱梯度作为优化的色谱条件,并确定各酚类物质在优化条件下的保留时间,完成目标物的定性。
1.2.5 混合标准储备液的配制 称取各种标准品适量,配制成高浓度母液,再用移液枪移取适量母液于100 mL 容量瓶,用去离子水定容至刻线,得到绿原酸300 mg/L、阿魏酸50 mg/L、虎杖苷50 mg/L、芦丁500 mg/L、水杨酸250 mg/L、反式白藜芦醇50 mg/L、槲皮素50 mg/L、山奈酚50 mg/L 的混合标准储备液;称取6 mg 矢车菊素-3-O-葡萄糖苷于1.5 mL 的样品瓶中,加去离子水配制成浓度为6000 mg/L 矢车菊素-3-O-葡萄糖苷储备液。
1.2.6 标准曲线的建立 依次分别吸取0.1、0.2、0.4、0.8、1.6、3.2 mL 混合标准储备液以及5、10、20、40、80、160 μL 矢车菊素-3-O-葡萄糖苷储备液于6 个10 mL 容量瓶中,用去离子水定容,得到6 个混合标样的浓度梯度。在优化的条件下对浓度梯度1~6 由低到高进行检测,将峰面积(Y)和浓度(x)进行强制过原点的线性拟合,建立标准曲线。
1.2.7 定量方法的方法学评估 把最低浓度标准溶液逐步稀释检测,分别取信噪比等于3 和10 时对应分析物的浓度作为检出限和定量限。取标样梯度3 重复进样6 次,计算各酚类物质的相对标准偏差。取两份相同的毛葡萄果酒的样品,其中一份加入浓度梯度2、4、6 标准混合溶液,平行测定六次,计算出相应组分的加标回收率。
1.2.8 发酵过程酚类物质变化的跟踪 利用所建立的HPLC 检测方法对酚类物质在22 ℃和28 ℃发酵变化进行跟踪,分别取第0、3、6、9、12、15 d 的样品进行检测。样品经0.22 μm 针头滤膜过滤后直接上样检测,每个温度的两个平行发酵均进行跟踪,每个取样点的样品进行三次重复测定。
1.3 数据处理
使用Microsoft Excel 2016 软件对数据进行处理,数据以两次实验的六个检测数据的平均值±标准偏差形式表示。
2 结果与分析
2.1 HPLC 色谱条件的优化
对12 mg/L 的矢车菊素-3-O-葡萄糖苷、12 mg/L的绿原酸、2 mg/L 的阿魏酸、2 mg/L 的虎杖苷、20 mg/L 的芦丁、10 mg/L 的水杨酸、2 mg/L 反式白藜芦醇、2 mg/L 槲皮素、2 mg/L 山奈酚的混标按线性洗脱梯度三进行了270~400 nm 的扫描,结果如图1 所示。矢车菊素-3-O-葡萄糖苷、绿原酸、阿魏酸、虎杖苷、芦丁、水杨酸、反式白藜芦醇、槲皮素、山奈酚检测的最大吸收波长分别是279、324、322、317、353、299、317、364、364 nm。为简化定量过程,选定定量波长依次为280、320、320、320、360、306、320、360、360 nm。
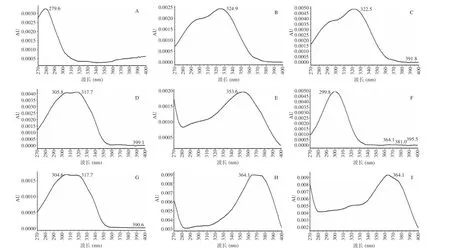
图1 九种酚类物质标样270~400 nm 的HPLC 二极管阵列光谱图Fig.1 HPLC diode array spectrograms of nine phenols at 270~400 nm
表1 列出的3 个有代表性的洗脱梯度对应的洗脱效果见图2。由于各物质在280 nm 均有检出,色谱峰分离效果均以280 nm 色谱图说明。采用梯度一洗脱时,矢车菊素-3-O-葡萄糖苷(峰1)与绿原酸(峰2)以及阿魏酸(峰3)、虎杖苷(峰4)与芦丁(峰5)均未完全分开(见图2A)。采用洗脱梯度二时,阿魏酸(峰3)、虎杖苷(峰4)与芦丁(峰5)已完全分离,但矢车菊素-3-O-葡萄糖苷与绿原酸分离效果仍不理想,二者的分离度仅为1.17(见图2B)。为此,将乙腈浓度达到16%的时间由28 min 改为梯度三的26 min,使二者的分离度达到1.53,实现了完全分离(见图2C)。另外,梯度三使最后出峰的山奈酚的保留时间由56.1 min 提前到53.3 min,提高了检测效率。最终确定梯度三为最优梯度。
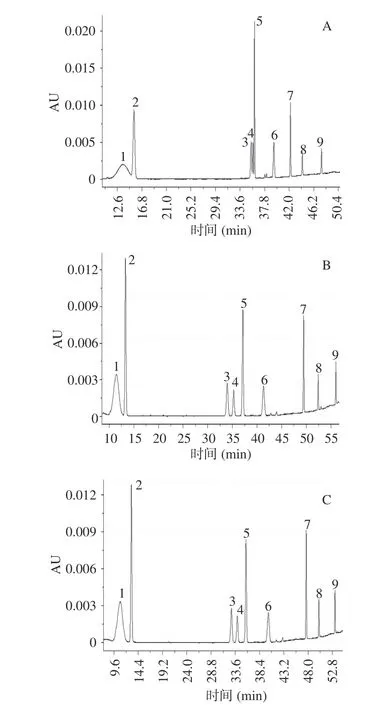
图2 九种酚类物质标样三个梯度下的280 nm HPLC 色谱图Fig.2 HPLC chromatograms of 9 phenolic compound standards at 280 nm under three gradients
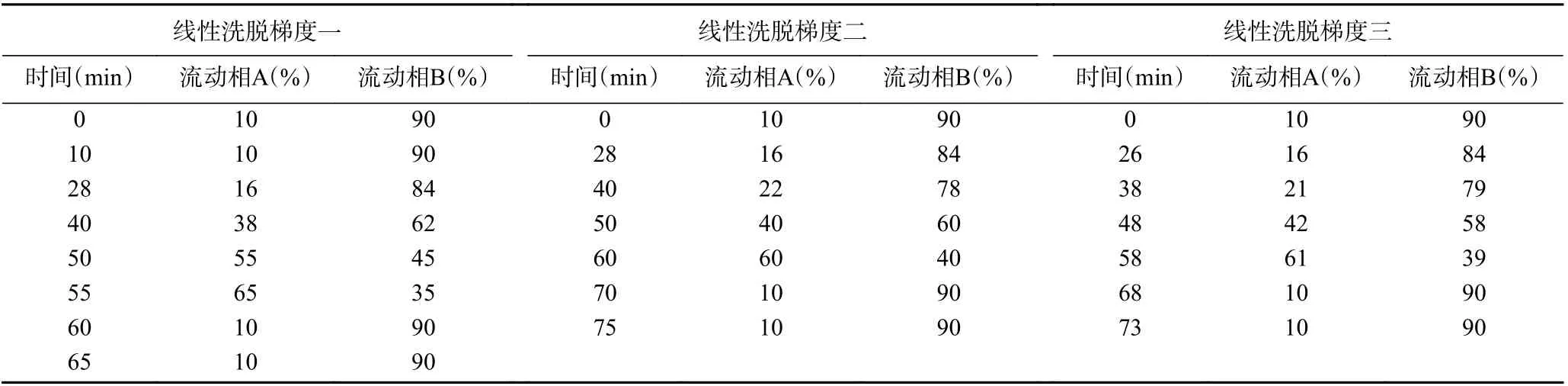
表1 优化过程所采用的三种洗脱梯度Table 1 The comparison of the three eluent gradients adopted in the optimization process
2.2 9 种酚类物质的定性
在优化的条件下,进行完成9 种酚类物质的定性,在4 个检测波长下的色谱图见图3,确定的保留时间见表2。

图3 优化条件下9 种混合标准品在四个检测波长的HPLC 谱图Fig.3 Chromatograms of 9 phenolic compounds at 4 detected wavelengths under optimum conditions
2.3 标准曲线建立及方法的评价
在优化的条件下对6 个混合标准梯度进行检测,将峰面积(y)和浓度(x)进行强制过原点的线性拟合,建立标准曲线,进行方法评价。标准曲线及其线性范围与R2、检出限、定量限、加标回收率、相对标准偏差的结果见表2。结果显示,该方法的线性范围较宽,标准曲线线性相关系数0.989~0.999;检出限0.017~0.296 mg/L,定量限0.056~0.987 mg/L,混合标样梯度4 对应的加标回收率92.37%~103.45%,相对标准偏差0.25%~3.26%,说明该方法线性良好,精密度和准确度高,可用于葡萄酒发酵液的检测。

表2 九种酚类物质的保留时间、检测波长及定量方法的评价Table 2 Evaluation of retention time,measure wavelength and quantitative methods of 9 phenolic compounds
2.4 发酵过程酚类物质的变化
温度对发酵的剧烈程度有明显的影响[21],利用所建立的HPLC 检测方法对酚类物质在22 ℃和28 ℃发酵15 d 的发酵液中变化进行跟踪,以期明确较大的温度差异对酚类物质的影响。为使表述更清晰,将所测9 种酚类物质按结构分类顺序进行讨论,即酚酸类(绿原酸、阿魏酸、水杨酸)、花色苷类(矢车菊素-3-O-葡萄糖苷)、芪类物质(反式白藜芦醇、虎杖苷)、黄酮醇类(山奈酚、槲皮素、芦丁)的顺序进行讨论[5]。
2.4.1 绿原酸、阿魏酸、水杨酸的变化 绿原酸属于对羟基苯甲酸型酚酸,水杨酸和阿魏酸属于对羟基肉桂酸型酚酸[5],三者的变化趋势不一致(见图4)。水杨酸呈现先降后升然后缓慢趋稳的趋势;绿原酸一直呈下降趋势;阿魏酸呈先升后降的趋势。葡萄浆果中酚酸以游离和结合两种形式存在,其中游离态占20%~25%[15],果肉中游离酚酸的含量高于果皮中[30]。三者不一致的变化趋势说明不同酚酸物质在发酵过程中经历了不同的浸提、水解、加合、转化等过程。温度对发酵过程略有影响,但15 d 时两个温度发酵液的水杨酸、绿原酸含量差异不大。15 d 发酵液中水杨酸含量约是魏巍等所报道的“桂葡1 号”葡萄酒的三分之一[28],可能与二者所采用的皮渣分离时间、酵母、葡萄的农事等差异有关。
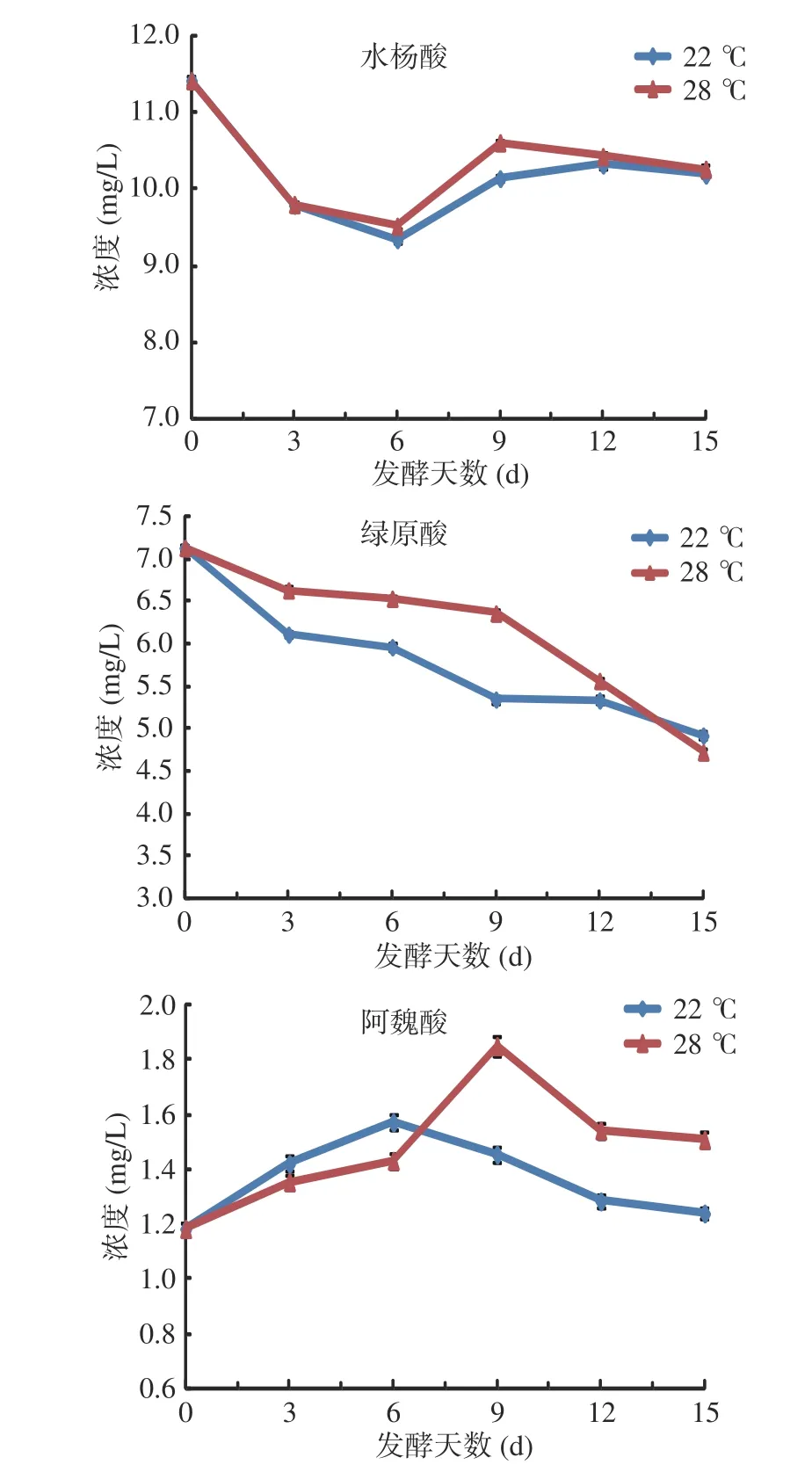
图4 发酵过程水杨酸、绿原酸、阿魏酸的含量变化Fig.4 Content change of salicylic acid, chlorogenic acid,ferulic acid during fermentation
2.4.2 矢车菊素-3-O-葡萄糖苷的变化 葡萄酒中的花色苷种类很多,包括基本花色苷、酰化花色苷、吡喃花色苷和聚合花色苷[12],矢车菊素-3-O-葡萄糖苷是葡萄酒中含量丰富的基本型花色苷[24]。矢车菊素-3-O-葡萄糖苷的变化见图5,呈先升后降趋势,与文献报道一致[15]。前期上升主要是浸提的结果,而后期下降可能是发生了水解、聚合或加合反应(如吡喃花色苷Vitisins 生成)[15,22,26]。
由图5 可知,不同发酵温度对矢车菊素-3-O-葡萄糖苷的变化以及发酵结束时的含量影响较明显。28 ℃的峰值高但后期下降也明显,在发酵结束时比22 ℃含量低。相对于初始发酵液,15 d 发酵结束时22、28 ℃的含量分别提高了109%和92%。发酵过程矢车菊素-3-O-葡萄糖苷呈增加趋势,与花色苷是葡萄酒的主要颜色来源的观点相符[12]。

图5 发酵过程矢车菊素-3-O-葡萄糖苷的含量变化Fig.5 Content change of cyandin-3-O-glucoside during fermentation
2.4.3 反式白藜芦醇与虎杖苷的变化 反式白藜芦醇和虎杖苷是葡萄酒中主要芪类物质,变化趋势见图6。由图6 可知,反式白藜芦醇在发酵过程呈先升后降的趋势;两个发酵温度前期差别不大,均在发酵第6 d 到达峰值,后期均下降,28 ℃的降幅相对较大;其15 d 发酵液的含量低于初始发酵液。发酵液中反式白藜芦醇的下降可能与酵母的吸附、对应糖苷的分解以及顺反异构的互相转化有关[20−21]。虎杖苷在发酵过程中一直下降,可能与酶促水解作用较强烈有关[20],28 ℃发酵液的前期降幅更明显。
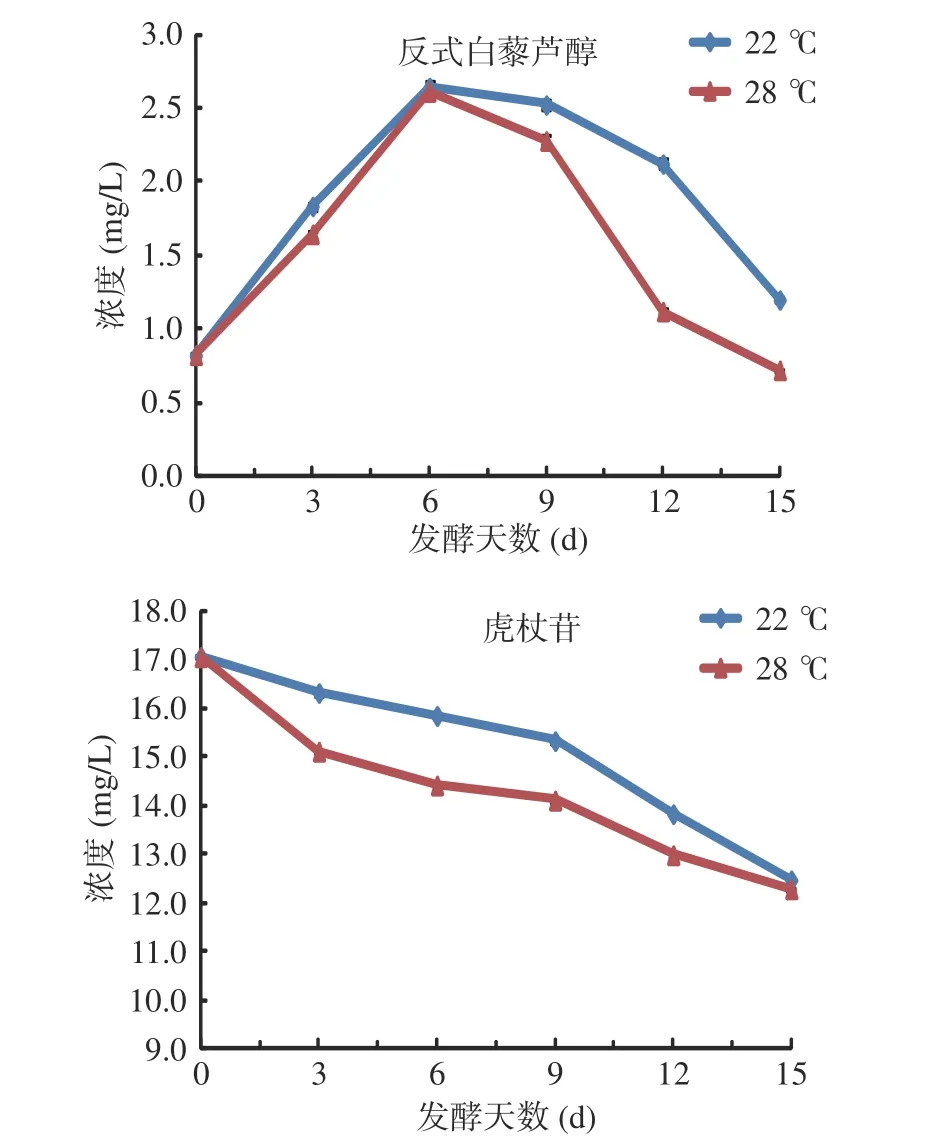
图6 发酵过程反式白藜芦醇与虎杖苷的含量变化Fig.6 Content change of trans-resveratrol and polydatin during fermentation
2.4.4 山奈酚、槲皮素、芦丁的变化 山奈酚、槲皮素、芦丁是葡萄酒中主要的黄酮醇类物质,发酵过程中山奈酚和槲皮素呈现先升后降再趋稳的变化(见图7),峰值出现在第6 d,相同发酵时间28 ℃发酵液的含量均高于22 ℃发酵液的含量。山奈酚、槲皮素的前期增加可能与酵母分泌的β-葡萄糖苷酶引发对应的糖苷水解有关[20]。芦丁也呈先升后降趋势(见图7),峰值在第3 d,其前期的增加可能与浸提有关,后期的下降可能是被糖苷酶水解的原因[20]。15 d发酵液中芦丁和槲皮素含量明显低于魏巍的报道水平[28],可能与所采用的原料、酵母、工艺的不同有关。

图7 发酵过程山奈酚、槲皮素、芦丁的含量变化Fig.7 Content change of kaempferol, quercetin, rutin during fermentation
3 结论
利用反相高效液相-紫外二极管阵列法建立了9 种酚类物质的定量分析方法。该方法的线性范围宽、检出限低、加标回收率高、精密度好。对“桂葡1 号”毛葡萄在22 ℃和28 ℃发酵过程中的酚类变化进行了跟踪,确定了不同酚类物质的变化规律。除虎杖苷和绿原酸一直呈下降趋势外,其它酚类物质的变化均呈先升后降的趋势。两个发酵温度的变化趋势基本一致,只是幅度不同。在符合先升后降的酚类物质中,除矢车菊素-3-O-葡萄糖苷外,两个温度的发酵曲线中峰值高的温度对应的15 d 发酵液中相应物质含量也高。
本研究为葡萄酒典型酚类物质的检测和大生产品质控制提供了一个有效手段,酿造过程的探讨为毛葡萄酒的优质化生产提供了前期的数据。但鉴于酚类物质的复杂性和影响因素的多变性,尚需要借助液质等先进手段对毛葡萄酒中的酚类物质进行深入的探究。
