内嵌金属碳氮化物团簇富勒烯的稳定性与生成机理
2021-11-22施俊杰胡子琦杨逸豪步宇翔施祖进
施俊杰,胡子琦,杨逸豪,步宇翔,*,施祖进,*
1山东大学化学与化工学院,济南 250100
2北京大学化学与分子工程学院,稀土材料化学及应用国家重点实验室,北京分子科学国家研究中心,北京 100871
1 引言
富勒烯分子具有纳米级别的空腔,在其中可以嵌入各种不同大小的金属或金属原子簇,形成具有独特核壳结构的富勒烯金属包合物1–3。利用内嵌金属与富勒烯碳笼之间的相互作用,可以调控富勒烯金属包合物的物理和化学性质。与空心富勒烯相比,富勒烯金属包合物表现出更为优异的性能,其化学反应性质与空心富勒烯也有很大的不同4,5。富勒烯金属包合物具有许多新奇的光、电和磁学等方面性质,在光电材料、生物医学和量子信息存储等领域有非常广阔的应用前景6–12。
对新结构富勒烯金属包合物的探索一直以来是富勒烯领域中的研究重点。新结构的出现不但能丰富富勒烯家族的种类,同时还有可能带来新的性质,拓宽包合物的应用前景。内嵌金属团簇富勒烯是富勒烯家族中一个重要的分支。很多具有不稳定结构的团簇能在富勒烯碳笼中稳定存在,这为研究纳米尺度下原子的独特行为提供了一个很好的模型。例如,M3N@Ih-C80(M = Sc, Y,etc.)具有很高的稳定性,是目前研究最多的一类内嵌团簇富勒烯13–15。理论研究表明,Ih-C80在接受内嵌团簇转移的六个电子后,表现出很大的HOMOLUMO能级差16。在Ih-C80碳笼中,还可以嵌入多于四个原子的特殊团簇,如Sc3C217,Sc4O218,Sc4O319,Sc3CH20,Sc3NC21等。这些团簇无一例外均向碳笼转移六个电子,验证了的特殊稳定性。
目前内嵌多原子大团簇富勒烯的研究通常局限于小半径的Sc原子和异常稳定的Ih-C80碳笼。寻找具有新结构的内嵌大团簇富勒烯是一个具有挑战性的课题。本论文从内嵌团簇与主体碳笼之间尺寸匹配的角度出发,通过量子化学计算探索了由更大的稀土金属原子组成的团簇与大碳笼富勒烯形成的包合物的稳定性。对于主体碳笼,我们选择了D2(186)-C96和D2(35)-C88(括号中的数字为异构体编号),它们接受六个电子形成的负离子和具有很高的稳定性,这一点与Ih-C80类似。对于内嵌的金属原子,我们选择了比Sc更大的Y、La和Gd等金属,研究对象为基于这几种金属的M3NC团簇嵌入D2(186)-C96和D2(35)-C88所形成的包合物。目前人们对于富勒烯及富勒烯包合物生成机理的认识尚不十分清楚,我们在分析富勒烯结构的基础上,提出了D2(35)-C88和D2(186)-C96这两种富勒烯之间的转变路径,对富勒烯的生成机理提供了新的认识,同时有助于加深对分子结构稳定性的理解。
2 计算方法
本研究中的量子化学计算在Gaussian 0922程序上完成。采用B3LYP泛函23对富勒烯金属包合物的结构进行优化。在初步优化时,对碳原子和氮原子采用3-21g(d)基组,对金属原子采用含有相对论效应校正赝势的CEP-4G基组。在此方法优化完成的基础上,再利用更高水平基组,即对碳原子和氮原子采用6-31g(d)基组,对金属原子采用SDD基组,进行二次优化得到精确结构。对优化以后的分子结构通过计算振动频率证明已达到能量极小点。利用GaussView软件查看前线分子轨道的空间分布图。
3 结果与讨论
3.1 M3NC@D2(186)-C96的稳定性
对于C96富勒烯碳笼,文献中已有报道在实验上可以得到La3N@C9624。为了研究M3NC@D2(186)-C96的稳定性,我们将其与相应的内嵌金属氮化物富勒烯进行了对比。采用密度泛函理论方法优化之后的M3NC@D2(186)-C96和M3N@D2(186)-C96(M = Gd, La)的分子结构如图1所示。在Gd3NC@D2(186)-C96和La3NC@D2(186)-C96分子中,Gd3NC和La3NC团簇均呈平面型,这与Sc3NC@C80中Sc3NC的平面构型类似。与La3N@D2(186)-C96分子相比,La3NC@D2(186)-C96分子中增加的C原子使得La―N―La的键角从125.7°增大至150.0°,同时La与碳笼上最近的C原子之间的距离从0.266 nm 缩短至0.258 nm。与Gd3N@D2(186)-C96分子相比,Gd3NC@D2(186)-C96分子中Gd―N―Gd键角由124.4°增大至135°,最近的Gd-C间距由0.257 nm缩短至0.252 nm。可见,Gd金属包合物的内嵌团簇在构型上的变化更小,证明了Gd3NC比La3NC更适合嵌入D2(186)-C96碳笼中,而La3NC需要有较大的结构变化才能保持平面构型。
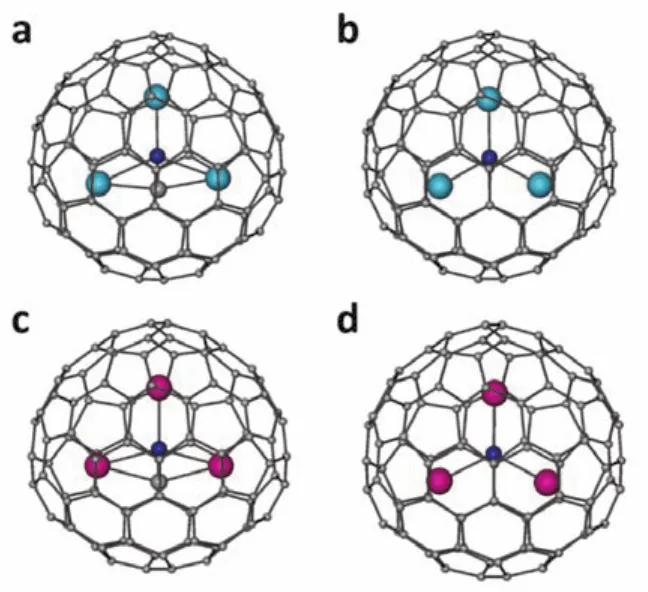
图1 (a)Gd3NC@C96,(b)Gd3N@C96,(c)La3NC@C96和(d)La3N@C96优化后的分子结构Fig. 1 DFT-optimized structures of (a)Gd3NC@C96,(b)Gd3N@C96, (c)La3NC@C96 and (d)La3N@C96.
为了更好地理解基于D2(186)-C96的金属包合物的稳定性,我们对该富勒烯及金属包合物进行了前线分子轨道分析。图2所示为D2(186)-C96、Gd3N@C96和Gd3NC@C96的分子轨道能级图。从图中可以看出D2(186)-C96具有很小的最高已占轨道-最低未占轨道(HOMO-LUMO)能级差(0.50 eV),因此D2(186)-C96非常不稳定。值得注意的是它的(LUMO + 2)-(LUMO + 3)能级差很大,所以D2(186)-C96容易接受六个电子形成稳定的。进一步分析表明,内嵌的Gd3NC和Gd3N团簇分别转移六个电子填充在了C96的三个未占轨道上,形成的两种富勒烯金属包合物均具有较大的HOMO-LUMO能级差(1.94和1.95 eV)。Gd3N@C96和Gd3NC@C96分子的HOMO和LUMO均为碳笼轨道主导,但是Gd3NC@C96的HOMO − 1轨道却主要分布在内嵌团簇上。这个团簇轨道的形状与文献中报道过的Sc3NC@C8021和Lu3C2@C8825的LUMO轨道形状一致,不同的是该轨道在Gd3NC@C96中的能级更低。通过轨道形状和轨道成分分析可知,该团簇轨道是由NC3−基团的π*反键轨道与Gd的d轨道相互作用而形成。Gd3NC@C96内嵌团簇中C原子与N原子之间的距离为0.128 nm,与Sc3NC@C80中C=N双键键长类似26,因此在Gd3NC@C96中C原子与N原子形成的化学键应为双键。C=N基团作为整体所呈现的电荷为−3,则氮原子的价态为−3,碳原子的价态为0。
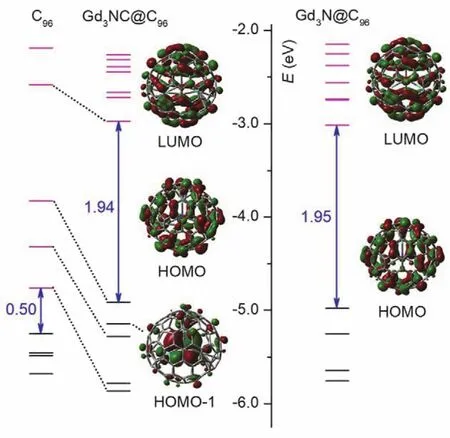
图2 C96、Gd3N@C96和Gd3NC@C96的前线分子轨道图Fig. 2 Molecular orbital energy diagram for C96,Gd3N@C96 and Gd3NC@C96.
3.2 M3NC@D2(35)-C88的稳定性
D2(35)-C88碳笼比D2(186)-C96的尺寸小,因此对于内嵌金属原子种类我们选取了Gd和离子半径更小的Y进行研究。采用密度泛函理论优化之后的M3NC@D2(35)-C88和M3N@D2(35)-C88(M = Gd, Y)结构如图3所示。Gd3NC和Y3NC团簇在D2(35)-C88碳笼内部均为平面构型。由于Gd3+的离子半径较大,Gd3NC团簇中Gd―N―Gd键角较大,为138.3°,与碳笼上最近的C原子之间的距离仅为0.244 nm。与Gd3NC@D2(186)-C96相比,Gd3NC团簇在D2(35)-C88碳笼中发生了更大的形变。而Y3+离子半径较小,Y3NC团簇与D2(35)-C88碳笼的尺寸更加匹配。图4所示为Y3N@C88和Y3NC@C88的前线分子轨道空间分布图。从图中可以看出Y3N@C88的HOMO和LUMO均分布在碳笼上。Y3NC@C88的HOMO与Y3N@C88的HOMO非常相似,但Y3NC@C88的LUMO既有碳笼轨道的成分,又有内嵌团簇的贡献,说明Y3NC团簇对分子的电子结构和化学性质有直接影响。
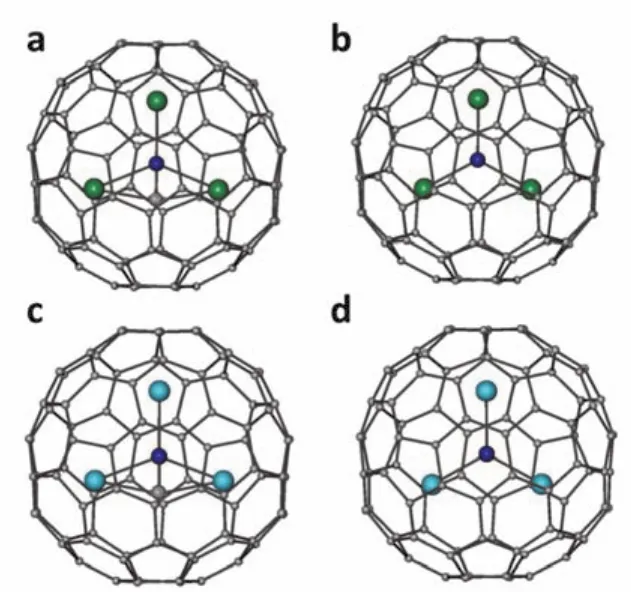
图3 (a)Y3NC@C88,(b)Y3N@C88,(c)Gd3NC@C88 和(d)Gd3N@C88 DFT优化后的分子结构Fig. 3 DFT-optimized structures of (a)Y3NC@C88,(b)Y3N@C88, (c)Gd3NC@C88 and (d)Gd3N@C88.
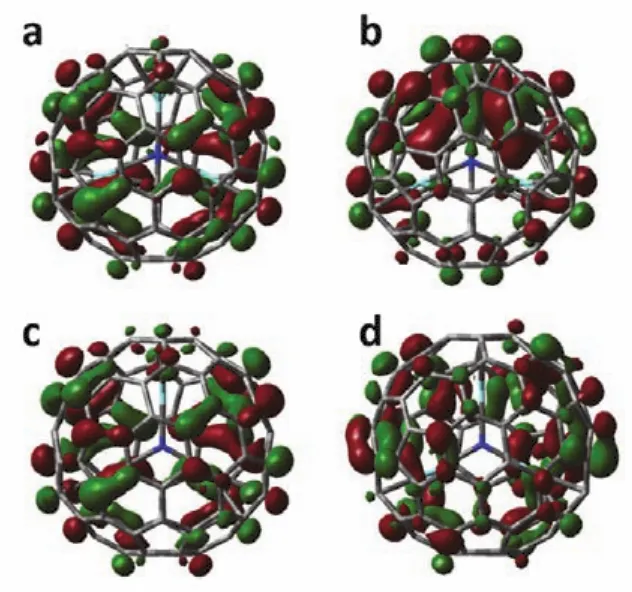
图4 Y3NC@C88的(a)HOMO和(b)LUMO,以及Y3N@C88的(c)HOMO和(d)LUMO轨道图Fig. 4 Spatial distribution of molecular orbitals for(a)HOMO and (b)LUMO of Y3NC@C88;(c)HOMO and (d)LUMO of Y3N@C88.
3.3 内嵌团簇与富勒烯的结合能
为了更好地理解内嵌团簇与富勒烯碳笼主体之间的尺寸匹配效应和选择性,我们计算了富勒碳笼和内嵌团簇之间的结合能,即单独团簇和空心富勒烯的能量之和减去富勒烯金属包合物的能量。结合能越大说明团簇与富勒烯碳笼之间的相互作用越强,包合物也越稳定。表1列出了计算得到的各种金属富勒烯包合物的结合能。从表中可以看出,对于D2(186)-C96碳笼,La3N比La3NC团簇嵌入的结合能更大,而Gd3N比Gd3NC团簇嵌入的结合能更小。因此,在实验上La3N@C96比La3NC@C96更易得到,而Gd3NC@C96比Gd3N@C96更加稳定。这与前述通过内嵌团簇变形程度得到的结论一致。对于D2(35)-C88碳笼, Y3NC与Y3N内嵌的结合能非常接近,说明二者都可以稳定存在;而Gd3NC比Gd3N内嵌的结合能小很多,说明在实验上更易获得Gd3N@C88。综合上述结果,可以看出随着Y、Gd、La离子半径和M3NC团簇尺寸的增大,需要有尺寸更大的碳笼与之匹配,从而形成稳定结构。
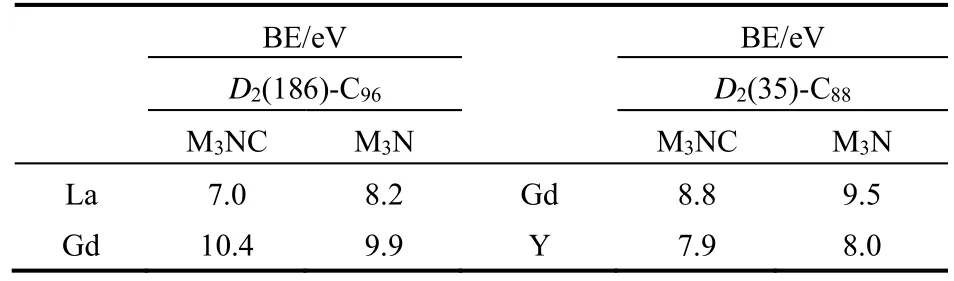
表1 D2(35)-C88和D2(186)-C96碳笼内嵌M3N和M3NC团簇时的结合能数值(BE)Table 1 Binding energy (BE)for encapsulation of M3N and M3NC clusters into D2(35)-C88 and D2(186)-C96 cages.
3.4 生成机理
除了本文研究的D2(186)-C96和D2(35)-C88富勒烯以外,Ih(7)-C80可以内嵌Sc3NC团簇形成稳定的包合物21。我们发现上述三种富勒烯碳笼的结构具有相似性。从D2(186)-C96、D2(35)-C88和Ih(7)-C80的Schlegel投影图(图5)可以看出,这三种富勒烯结构中的五元环都呈现出均匀的排布方式。它们的负离子都具有很高的稳定性,在尺寸匹配的基础上,它们都可以内嵌氮化物M3N和碳氮化物M3NC团簇形成稳定的包合物。
在富勒烯的生长机理研究中,研究者们一般持有两种观点,即“自上而下”和“自下而上”两种机理27–30。富勒烯碳笼失去偶数个碳原子形成更小的富勒烯是一种“自上而下”的富勒烯生成机理,而其逆过程是一种“自下而上”的机理。这两种机理可能共存。我们对D2(186)-C96和D2(35)-C88二者之间的转变路径进行了研究,下面以“自下而上”的方式进行描述。
在图6a中所示的初始结构中三个五元环为均一排布,它可以通过两步增加C2的路径发生重排,最终得到的结构仍具有五元环均匀分布的特点,保持了结构的稳定性。这两步增加C2的过程分别称为R1和R2过程。此外,在富勒烯生成过程中还有可能发生图6b所示的Stone-Wales异构化转变,记为R3过程。目前已报道过的富勒烯生成机理大多涉及R1和R3过程,而对R2的研究还很少。同时,R3即Stone-Wales异构化过程通常需要很大的能垒31。因此我们以R1和R2转变过程为出发点,对D2(186)-C96和D2(35)-C88的结构进行了详细的研究,提出了它们之间的转变路径,如图7所示。具体来说,以D2(35)-C88为初始结构,通过两次R1和R2转变的组合,以C1(99832)-C90、C1(66)-C92和C1(150347)-C94为中间体,最终可以转化为D2(186)-C96。在初始结构和最终结构中,五元环均为均匀排布,因此保持了结构的稳定性。R1转变过程的能垒大约在293–334 kJ·mol−1,在高温下有可能实现。
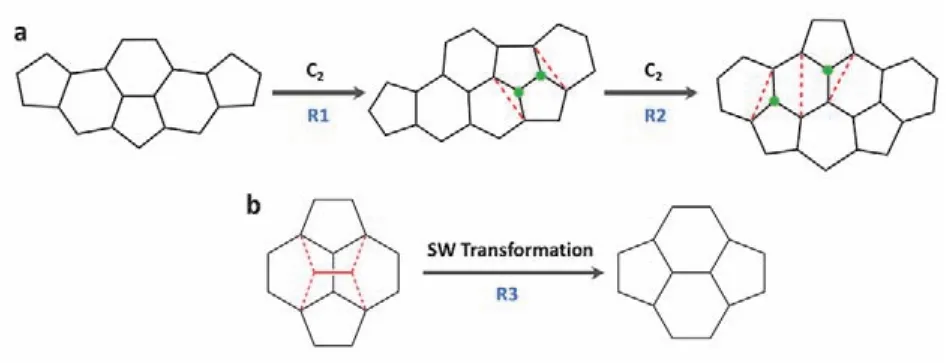
图6 (a)五元环均匀分布结构的重排机理;(b)Stone-Wales异构化过程Fig. 6 (a)Rearrangement mechanism;(b)Stone-Wales transformation.
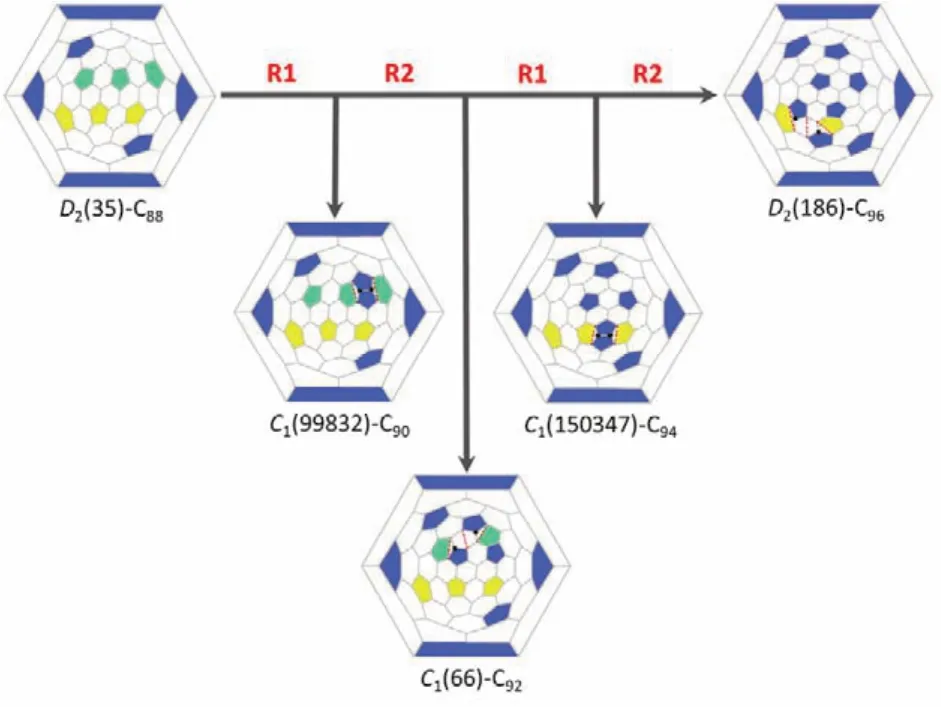
图7 D2(186)-C96到D2(35)-C88结构转变示意图Fig. 7 Transformation process from D2(186)-C96 to D2(35)-C88.
4 结论
我们对内嵌M3NC团簇富勒烯的稳定性、结构及相互之间的联系进行了量子化学计算研究。通过与相对应的内嵌氮化物团簇富勒烯进行对比,阐明了碳氮化物团簇与主体碳笼之间的尺寸匹配效应。同时,以偶数碳原子的增加为基础,我们提出了不含Stone-Wales异构化的富勒烯碳笼生成路径,在C88和C96之间建立了结构联系,并为新结构的出现提供了理论支持。
猜你喜欢
杂志排行

物理化学学报的其它文章
- Hollow Nitrogen-Rich Carbon Nanoworms with High Activity for Metal-Free Selective Aerobic Oxidation of Benzyl Alcohol
- Photocrosslinking-Immobilized Polymer Vesicles for Lowering Temperature Triggered Drug Release
- CO Hydrogenation to Ethanol over Supported Rh-Based Catalyst:Effect of the Support
- CdTeSe合金幻数团簇的室温合成和形成机理研究
- 体相界面导通的复合快离子导体
- 异氰酸苯酯诱导的类胶原多肽自组装