CO Hydrogenation to Ethanol over Supported Rh-Based Catalyst:Effect of the Support
2021-11-22ZilongShaoXiaofangLiuShunanZhangHuiWangYuhanSun
Zilong Shao , Xiaofang Liu , Shunan Zhang , Hui Wang ,*, Yuhan Sun ,3,*
1 CAS Key Lab of Low-Carbon Conversion Science and Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, China.
2 University of Chinese Academy of Science, Beijing 100049, China.
3 School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China
Abstract:Ethanol has great application prospects given it is an important essential chemical and a substitute for traditional energy sources. Currently,ethanol production is achieved through grain fermentation and petroleum-based ethylene hydration. However, the inefficient fermentation processes and increasingly depleted crude oil resources hinder the large-scale production of ethanol. Therefore, the development of alternative technologies for ethanol production has become an important issue. The direct production of ethanol from syngas (CO + H2)is considered to be a new strategy to acquire high value-added products and achieve clean utilization of carbonaceous resources such as coal,natural gas, and biomass. Supported Rh-based catalysts have been extensively studied as the most promising and effective systems for the direct production of ethanol from syngas. The use of promoters and supports is generally effective in increasing the activity and ethanol selectivity of supported Rh-based catalysts. Fe is widely used in the research on Rh-based catalysts, as it is one of the most effective promoters for enhancing ethanol selectivity. In this work, with the aim of exploring the role of the support,we used the incipient wetness impregnation method to prepare Fe-promoted Rh-based catalysts supported by CeO2, ZrO2,and TiO2 for the synthesis of ethanol from syngas. CO conversion of CO on the RhFe/TiO2 catalyst was as high as 18.2% under the reaction conditions of 250 °C and 2 MPa, and the selectivity to ethanol in the alcohol distribution was 74.7%,which was much higher than that observed with RhFe/CeO2 and RhFe/ZrO2 under the same conditions. The characterization results showed that the specific surface of the catalyst followed the order RhFe/CeO2 < RhFe/ZrO2 Key Words:Rh-based catalyst; Support effect; Ethanol synthesis; Syngas conversion; Rh dispersion; Iron Ethanol is widely applied as potential fuel alternative and fuel additives in indispensable engine fuels1, and its production conventionally relies on the grain fermentation, which limits its production due to deficient grains2. Developing new routes for ethanol production with industrial potential is of great importance and direct conversion of syngas (a mixture of CO and H2)to higher alcohols (C2+OH, especially C2H5OH)is considered promising for clean and efficient use of carbonaceous resources such as coal, natural gas and biomass3–5. So far, four types of catalysts have been reported for such conversion and Rh-based ones have shown most effective6compared to other catalysts consisting of modified methanol (typically Cu-based)7,modified Fischer-Tropsch and Mo-based ones8. However,unpromoted Rh-based catalysts supported on common oxide such as SiO2or Al2O3showed relatively poor activity and ethanol selectivity9. Therefore, various promoters and supports have been developed for CO hydrogenation towards ethanol with high activity and selectivity. Research on the effect of promoters have been explored extensively in the past decades10–15. Fe has been studied as one of the most effective promoters of Rh-based catalysts for syngas to ethanol. It has shown that the addition of Fe inhibits the formation of hydrocarbon and greatly enhances the yield of C2+oxygenates, especially ethanol16. Although there is no consensus on the mechanism of Fe promotion due to different catalyst preparation methods and reaction conditions, some of the researchers believe that Fe could form a new active site with Rh for ethanol formation16,17. What's more, the physical and chemical properties of the support can influence the activity and selectivity of the catalyst remarkably18. For example, it has been found that the activity of the catalyst strongly depends on the surface area of the support19. In addition, support can also affect the reducibility of the loaded metal, due to the interaction between metal and support20. Metal oxide has been widely employed as support to form metal-support interaction21.Therefore, we attempted to study the effect of metal oxide support on CO hydrogenation to ethanol over Fe-promoted Rhbased catalyst. Herein, CeO2, ZrO2and TiO2have been used as supports for RhFe-based catalysts to investigate the catalytic performance for ethanol synthesis. The results show that better dispersion of Rh facilitates the production of more active sites and TiO2-supported catalysts with more O-vacancy have stronger CO adsorption capacity. Thus, RhFe/TiO2achieves 18.2% CO conversion as well as 74.7% ethanol selectivity in alcohol distribution. Such effects of support on performance of CO hydrogenation are investigated by N2adsorption-desorption, X-ray diffraction (XRD), H2-temperature programmed reduction(H2-TPR), X-ray photoelectron spectroscopy (XPS)and CO-temperature programmed desorption (CO-TPD). The monometallic Rh or Fe and bimetallic Rh-Fe catalysts were synthesizedviaIWI (incipient wetness impregnation)by the dissolution of rhodium (III) nitrate dehydrate(Rh(NO3)3∙2H2O, 98%, Energy Chemical)and iron (III)nitrate nonahydrate (Fe(NO3)3∙9H2O, 99.99%, SCRC)in distilled deionized water. The supports used in the experiment were commercial CeO2 (99.99%, SCRC), ZrO2 (99.99%, SCRC),TiO2(99.99%, Shanghai Powjer Industrial Co., Ltd). The resulting solution was added dropwise to 10 g of support (CeO2,ZrO2, TiO2)and continuously stirred at room temperature until a paste was formed, which was first dried at 40 °C for 6 h and then dried at 65 °C overnight. The catalyst calcined at 500 °C in air for 4 h and the heating rate is 5 °C∙min−1. Nominally, the contents of Rh and Fe were 2% (w, mass fraction)and 4% (w),respectively. The catalytic samples are labeled as follows:RhFe/CeO2, RhFe/ZrO2, RhFe/TiO2and Fe/TiO2. N2adsorption/desorption experiments were conducted at−196 °C on a TriStar II 3020. The catalysts degassed under vacuum conditions at 200 °C for 6 h prior to adsorption. The Brunauer-Emmett-Teller (BET)method used to determine the surface area. The total pore volume (TPV)calculated from the amount of vapor adsorbed at a relative pressure (p/p0)close to unity, wherepandp0are the measured and equilibrium pressures, respectively. The pore volume and pore size of the catalyst are calculated using the Barrett-Joyner-Halenda (BJH)model. X-ray powder diffraction (XRD)experiments executed using Rigaku Ultima 4 X-ray diffractometer utilizing Cu Kα radiation(40 kV, 40 mA)in the range of 5°–90° and scanning speed was 2 (°)∙min−1. Temperature programmed reduction (H2-TPR)was carried out in a Micromeritics Autochem-II 2920. Firstly, 0.05 g catalyst was first dried at 200 °C for 2 h to remove water under a He atmosphere. Then cooled down to 50 °C. Finally, 10% H2/Ar (50 mL∙min−1)was injected and the temperature was raised from 50 to 800 °C at the rate of 10 °C∙min−1. The off gas was analyzed by a thermal conductivity detector (TCD). X-ray photoelectron spectroscopy (XPS)were performed over a Thermo Scientific K-Alpha instrument equipped with AlKαradiation (12 kV, 4 mA,hν= 1486.6 eV)under ultrahigh vacuum(10−7Pa). Prior to each test, the calcined sample reduced in pure hydrogen at 350 °C for 6 h, and XPS measurements recorded with the exclusion of air contact after reduction. The binding energies were calibrated internally by adventitious carbon deposit C (1s)withEb= 284.8 eV (accuracy within ±0.1 eV). CO temperature-programmed desorption (CO-TPD)measurements performed in a Micromeritics Autochem-II 2920 with a thermal conductivity detector (TCD). 0.1 g of the sample wasin situreduced with pure 10% H2/Ar (50 mLmin−1)at 350 °C for 2 h. After cooling to 50 °C, the sample was flushed with He (40 mLmin−1)for 30 min at 50 °C, after which the sample exposed to pure CO (30 mLmin−1)for 1 h and then flushed with He flow (30 mLmin−1)to remove all physically adsorbed molecules. Desorption was then performed by increasing the temperature from 50 to 600 °C at a rate of 10 °C min−1under a flow of helium gas. The carbon monoxide hydrogenation reaction carried out in a 5 mL fixed bed stainless steel tubular reactor (800 mm in length and 8 mm in inner diameter). Prior to testing, 1.5 g of the catalyst diluted with 3.0 g of quartz sand (both in 40–60 mesh)reduced in a pure hydrogen stream (20 mL∙min−1)at 350 °C for 6 h under a pressure of 0.1 MPa. After reduction, the gas flow was switched to syngas (H2/CO = 2.0)with 40 mLmin−1.Subsequently, the reaction pressure gradually increased to the target value, and the temperature rose to the reaction temperature at a rate of 2 °C∙min−1. After passing through the hot trap and the cold trap in turn, the tail gas analyzed online by gas chromatography (GC). All gas phase products analyzed using two online gas chromatographs (GC). A TDX carbon molecular sieve column analyzed H2, N2, CO, CH4and CO2with a thermal conductivity detector (TCD)using Ar as a carrier gas.Hydrocarbons analyzed by modified alumina packed columns,where Ar used as a carrier gas and hydrogen ionization detector(FID). Off-line analysis of aqueous products collected from cold traps and hot traps by GC analysis of aqueous products by two Porapak Q columns equipped with TCD (for H2O and MeOH detection)and FID (for C1–C5oxygenate detection). The CO conversion and product selectivity were calculated and the specific calculation equations are as follows: WhereNiis the number of carbon atoms of the producti,Miis the molar concentration of producti. The performance of CO hydrogenation on catalysts with various supports was given in Fig. 1. As observed, RhFe/CeO2gave CO conversion as low as 5.1% and 25.6% of ROH selectivity with major methanol in alcohols. RhFe/ZrO2promoted the CO conversion rate to 7.2% and the selectivity of alcohol product to 27.8%, respectively. At the same time, the selectivity of methanol decreased, while ethanol increased in the alcohol distribution. Encouragingly, the highest CO conversion(18.2%), alcohol selectivity (33.2%)and ethanol selectivity(74.7%)in the alcohol distribution could be achieved over RhFe/TiO2, while the selectivity towards hydrocarbon products declined to 62.4%. CO2 selectivity over all catalysts was lower than 5%. In addition, the Fe/TiO2catalyst without Rh showed a conversion of 1.4% and almost no alcohol was produced under our experimental conditions (see Table S1 for details, in Supporting Information), which confirmed that Fe acted only as a promoter. Apparently, TiO2-supported catalyst surpassed CeO2- and ZrO2-supported ones in terms of CO conversion and ethanol selectivity, indicating that support had a significant effect on catalytic performance and various characterization analyses were conducted to clarify the effect of support, as described in the following section. Fig. 1 (a)CO conversion and product selectivity, (b)alcohol distribution. The X-ray diffraction patterns of samples after calcination had been depicted in Fig. 2. The diffraction peaks of RhFe/CeO2at 28.6°, 33.2°, 47.6°, and 56.5° were attributed to cerianite CeO2(JCPDS 34-0198). The typical diffraction peaks of monoclinic ZrO2 (JCPDS 83-0940)was easily observed at 28.3°, 31.5°,34.2° and 50.2° on RhFe/ZrO2. 80% anatase TiO2(JCPDS 89-4921)and 20% rutile TiO2(JCPDS 21-1276)at 25.5°, 38.1°,48.3° and 27.7°, 41.6° could be clearly confirmed for TiO2-supported sample. All catalysts exhibited none of diffraction peaks corresponding to Rh or Fe species due to their low contents and small particle size22. In addition, average crystallite size of the three supports estimated by the Scherrer's equation were 90.9, 35.9 and 24.4 nm, respectively. Fig. 2 XRD patterns of different catalysts. Table 1 listed textural properties of the samples detected by N2adsorption-desorption. RhFe/TiO2showed a specific surface area of 41.5 m2g−1, larger than RhFe/CeO2of 10.8 m2g−1and RhFe/ZrO2 of 13.1 m2g−1. In addition, RhFe/TiO2 sample possessed biggest pore volume and pore size. Therefore, it was expected that RhFe/TiO2catalyst with largest specific surface area was conducive to high dispersion of Rh species than the other two catalysts. The results of Rh dispersion and particle size for the RhFe-based catalysts were also tabulated in Table 1.Furthermore, the dispersion of Rh of the RhFe/TiO2 catalyst was 48.8%, which was higher than the others were. That meant Rh particles were better dispersed on TiO2support than ZrO2and CeO2, enhancing the contact interface between the highly dispersed Rh species and the Fe promoter, which was favorable for the formation of ethanol. On the other hand, the Rh particle size of RhFe/CeO2, RhFe/ZrO2 and RhFe/TiO2 were calculated to be 4.7, 3.7 and 1.9 nm, respectively. The Rh particle size could be affected by the support, which agreed with the conclusion reported by Kimet al.23. Table 1 Textural and structural properties, Rh dispersion and particle size of different supports. H2-TPR studies have been carried out and the profiles of all samples were shown in Fig. 3. For the RhFe/CeO2, a remarkable peak at 106 °C attributed to the reduction of Rh2O3 to Rh024.Two peaks at 209 °C and 301 °C corresponded to the reduction of Fe2O3to Fe3O4and Fe3O4to Fe0respectively. Moreover, the high temperature region around 720 °C on RhFe/CeO2was assigned to the massive reduction of Ce4+15. On RhFe/ZrO2sample, the reduction peak of Rh2O3appeared at 97 °C, and thatof iron oxide was between 209 °C and 301 °C. By contrast, the reduction of Rh2O3on RhFe/TiO2sample occurred at 65 °C. In addition, a broad peak emerged from 215 °C to 621 °C, which was considered to cover reduction peak of both iron oxide and the support, as TiO2 could be reduced above 250 °C previously reported by Hanet al.26. Moreover, according to H2consumption, not only iron oxide but also a portion of TiO2at 700 °C was reduced on Fe/TiO2. Fig. 3 H2-TPR profiles of the different catalysts. As can be seen from the above results, the reduction temperature of Rh2O3 sequentially declined in the following order: RhFe/CeO2 > RhFe/ZrO2 > RhFe/TiO2, which could be owed to the enhanced dispersion and decrease in particle size as demonstrated through H2chemisorbed26,27. Comparing with Fe/TiO2catalyst, the observed reduction peak of iron oxide shifted to the low temperature due to the existence of Rh species on RhFe/TiO2, which has been reported before that Rh could accelerate the reduction of iron oxide28. Furthermore, the TiO2 support was also reduced on RhFe/TiO2and the similar result on Rh-Cu/TiO2was reported by Stateet al.18. In general, H2-TPR results revealed the existence of possible interactions between Rh and support as well as Rh and Fe, which might be conducive to CO hydrogenation reaction. XPS were used to determine the chemical state and electronic properties of the surface elements. The XPS spectrum of the reduced samples was presented in Fig. 4, the corresponding binding energies (BEs)and surface atomic percentage were compiled in Table S2 (in Supporting Information). From Fig. 4a,BEs locating at 307.1–307.2 eV and 308.8–308.9 eV were attributed to Rh0and Rh+, respectively29. In addition, according to the H2consumption of the TPR curve, the rhodium oxide cannot be completely reduced. Therefore, it can be confirmed that both oxidized and metallic Rh species were present on the surface of the Rh catalyst after reduction. It was generally recognized that Rh0is the active site for CO dissociation, and Rh+facilitates the insertion of CO to form C2-oxygenates intermediates30,31. More Rh0as well as Rh+sites (see Table S2)on surface of catalyst might result in not only higher activity also selectivity towards alcohol products on RhFe/TiO2. Fig. 4 XPS analysis of different catalysts. The BEs of Fe 2p3/2in Fig. 4b were considered to the Feδ+(Fe3+or Fe2+)32. The promotion of the oxidation state of Fe has previously been reported16,33. These authors suggested Fe3+(or Fe2+)could increase the selectivity toward C2+ oxygenates.Wanget al. thought that the active site for the formation of C2-oxygenates was (Rhx0-Rhy+)-O-Mn+, wherein the M was the promoter which in close contact with Rh14. According to the foregoing results, Fe species could be more sufficiently contacted with Rh due to higher dispersion of Rh on the TiO2support. Based on this, we speculated that the reason for the increase of ethanol selectivity was that there were more (Rhx0-Rhy+)-O-Feδ+active sites. Hence, the highest ethanol selectivity appeared on RhFe/TiO2 catalyst since it contained the most active sites. In addition, O1s spectra in Fig. 4c were deconvoluted into three main peaks, which included surface lattice oxygen (OI),mobile oxygen ions (OII)like O2−or O−, and oxygen in hydroxyl groups (OIII)34. Table S3 (in Supporting Information)showed that the ratio of OII/(OI + OII + OIII)in RhFe/TiO2 was much higher than that of the others. As OIIwas closely correlated with the formation of O-vacancies34, a large number of oxygen vacancies formed over RhFe/TiO2, which might be attributed to easily reducible TiO2(see H2-TPR results). XPS spectra of the supports in Fig. 4d could demonstrate this assumption. The Ti 2pspectrum showed two different binding energies marked C and D, which were ascribed to Ti4+and Ti3+, respectively35. TiO2would be reduced after the reduction by H2, while the other two supports not (see Figs. S1 and S2, in Supporting Information),which was consistent with the conclusions obtained by H2-TPR. The CO-TPD technique was used to investigate the CO desorption behavior of various samples as showed in Fig. 5.Notably, the CO desorption peak intensity of RhFe/CeO2and RhFe/ZrO2were exceedingly weak compared with RhFe/TiO2.The difference in dispersion of Rh could account for it to some extent, but there must be another reason. As was known in the literature, both oxygen vacancies and Ti3+cations derived from readily reduced TiO2 could absorb CO36. H2-TPR and XPS results showed that partial reduction of TiO2could occur and generated Ti3+cations and a large amount of oxygen vacancies.Hence, highest dispersion of Rh, the maximum number of oxygen vacancies and the existence of Ti3+cations should be responsible for the strongest peak intensity of the RhFe/TiO2catalyst. Fig. 5 CO-TPD profiles of all catalysts. In addition, the CO desorption temperature increased gradually and followed the order RhFe/CeO2< RhFe/ZrO2 In summary, RhFe-based catalysts over various supports(CeO2, ZrO2and TiO2)have been employed to produce ethanol from syngas, and RhFe/TiO2proved to achieve best CO conversion (18.2%)and ethanol selectivity (74.7% in alcohols distribution). The characterization findings clearly demonstrated the effects of support, which lied in the following two aspects.Firstly, larger specific surface area on RhFe/TiO2 led to better Rh dispersion and smaller particle size, increasing the interface between well dispersed Rh and Fe, thereby enhanced the amount of (Rhx0-Rhy+)-O-Feδ+active site for ethanol synthesis over RhFe/TiO2based on H2-TPR and XPS results. Secondly, readily reducible TiO2 generated more oxygen vacancies and Ti3+cations, which increased the adsorption capacity of CO.Moreover, CO-TPD also indicated that the increment of CO adsorption strength in RhFe/TiO2compared with RhFe/CeO2and RhFe/ZrO2, which should be responsible for the high activity of the catalyst, thereby greatly enhancing the catalytic performance for CO hydrogenation to ethanol over RhFe/TiO2. Supporting Information:available free of chargeviathe internet at http://www.whxb.pku.edu.cn.1 Introduction
2 Experimental section
2.1 Catalyst preparation
2.2 Catalyst characterizations
2.3 Testing of the catalytic activity
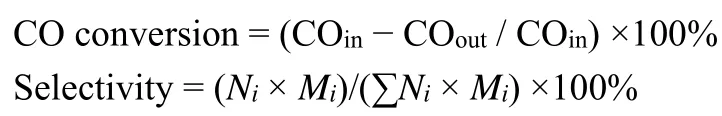
3 Results and discussion
3.1 Catalytic performance
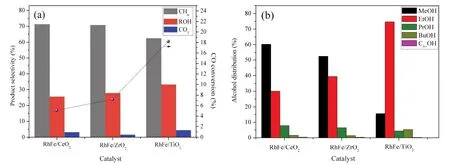
3.2 Catalyst structural properties
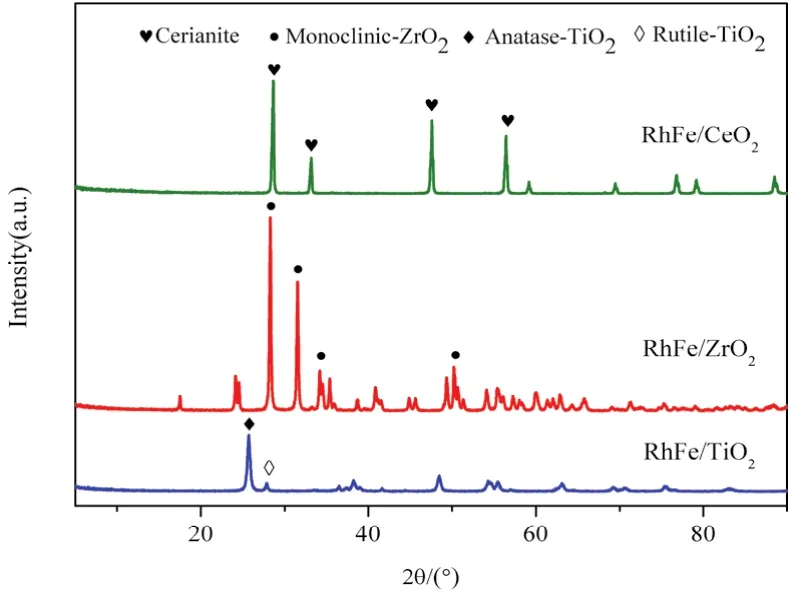


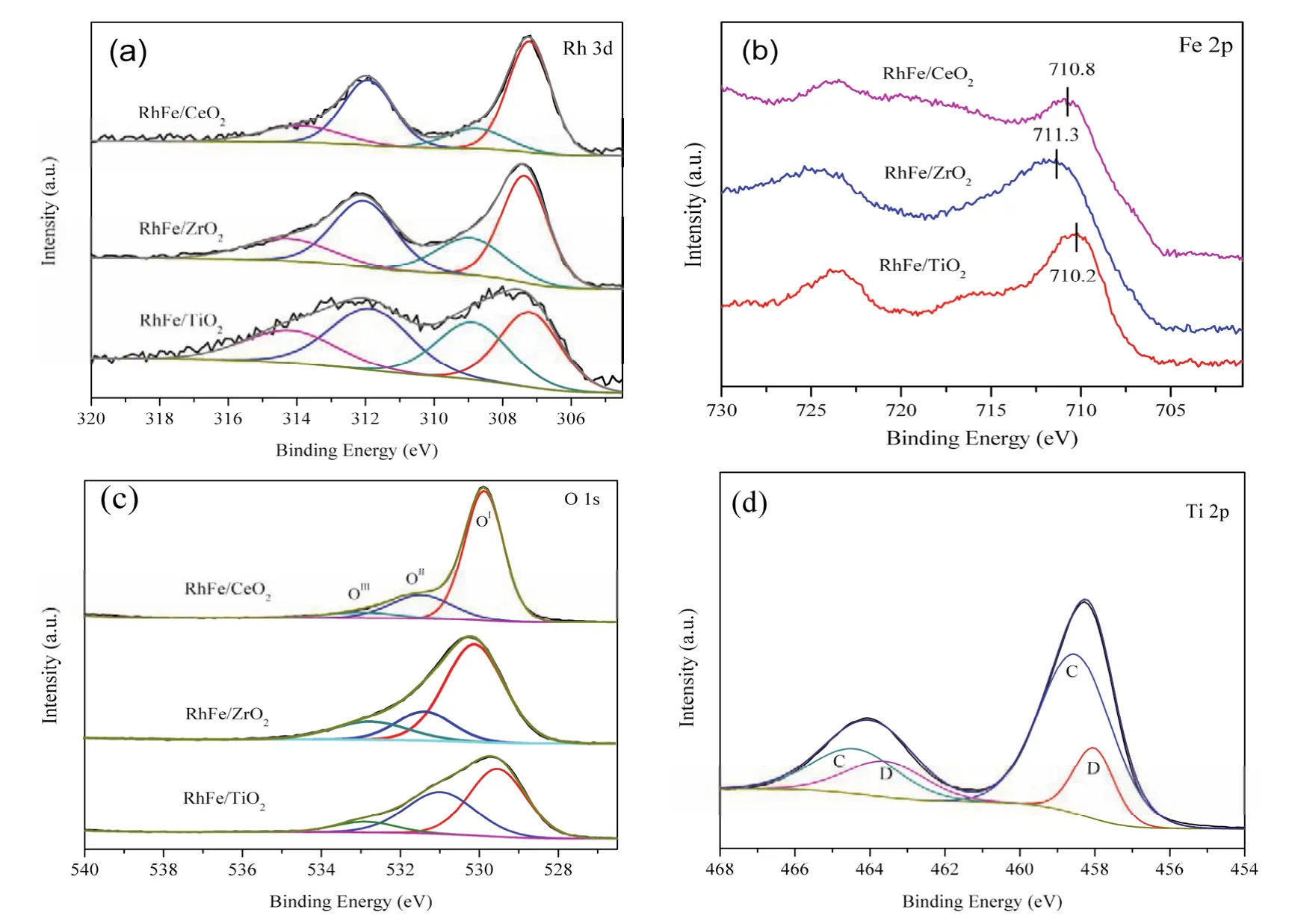

4 Conclusions
杂志排行

物理化学学报的其它文章
- Photocrosslinking-Immobilized Polymer Vesicles for Lowering Temperature Triggered Drug Release
- Poly(ε-caprolactone)-Polypeptide Copolymer Micelles Enhance the Antibacterial Activities of Antibiotics
- ReaxFF MD局部区域反应追踪与物理性质可视化分析
- 稀土-天然皮革可穿戴X射线防护材料的合成及性能
- 石墨烯玻璃透明薄膜加热特性
- Single-Molecule Field-Effect Transistors with Graphene Electrodes and Covalent Pyrazine Linkers