一测多评法同时测定九制大黄丸中5 种蒽醌类成分*
2021-11-21袁凤娟杨爱华
许 波,袁凤娟,杨爱华,郑 丽*
(1 江苏省南通市妇幼保健院药事科,南通 226018;2 新疆维吾尔自治区乌鲁木齐市第一人民医院分院药剂科)
一测多评法(quantitative analysis of multi-components by a single marker,QAWS)是指以样品中的某一成分(对照品易得且稳定)为内参物,建立该成分与其他成分的相对校正因子(relative correction factor,RCF),通过RCF 计算其他成分的含量,适用于对照品获得困难情况下的多成分同时测定[1]。九制大黄丸是以大黄药材九蒸九晒加工而成,用于胃肠积滞所致的便秘、湿热下痢、口渴不休、停食停水、胸热心烦、小便赤黄、头痛目眩[2]。由于中药材中成分种类复杂,与测定单一成分相比,多指标测定的质量评价方法更符合中药特点[1,3]。多指标测定必须有足够多的对照品,但是由于有的对照品难以分离、单体不稳定及价格昂贵等原因,使其获得较困难[4-8]。QAWS 可在节约对照品的基础上准确有效地对药物进行质量控制。
1 材料与方法
1.1 仪器 Dionex u3000 高效液相色谱(high performance liquid chromatography,HPLC)仪(四元泵、自动进样器、TCC-3000 RS 柱温箱、DAD-3000 检测器、Chromeleon 7 工作站);Waters e2695 HPLC 仪(四元泵、自动进样器、脱气装置、2998PDA 检测器、Empower工作站);Symmetry C18(250 mm×4.6 mm,5 μm)色谱柱;Aglient TC-C8(250 mm×4.6 mm,5 μm)色谱柱;H.H.S4 电热恒温水浴锅;Willi-Q 超纯水系统;Willipore 公司SK5200H 超声波清洗器;Sartorius BT 25 S型电子天平。
1.2 对照品与样品 芦荟大黄素(批号:110795-201007,纯度98%)、大黄素甲醚(批号:110758-201013,纯度99.8%)(中国药品生物制品检定所),大黄酸(批号:110757-200206,纯度≥98%)、大黄素(批号:110756-200110,纯度≥98%)、大黄酚(批号:110796-201118,纯度99.5%)(中国食品药品检定研究院)、九制大黄丸(河南宛东药业有限公司)。
1.3 试剂 甲醇、乙腈均为色谱纯、三氯甲烷(国药集团化学试剂有限公司)、超纯水(自制)。
1.4 色谱条件 Symmetry C18(4.6 mm×250 mm,5 μm)色谱柱;流动相为甲醇(A)-0.1%磷酸溶液(B)梯度洗脱(0~10 min,5%~30%A;10~40 min,30%~60%A;40~60 min,60%A;60~70 min,60%~100%A;70~80 min,100%A),流速1 mL/min;柱温30 ℃;检测波长430 nm。
1.5 混合对照品溶液的制备 分别精密称取芦荟大黄素3.01 mg、大黄酸4.50 mg、大黄素4.09 mg、大黄素甲醚7.99 mg 置于20 mL 容量瓶中,精密称取大黄酚6.10 mg 置于10 mL 容量瓶中,加甲醇适量,使其溶解后加甲醇至刻度,摇匀,分别配置成5 个对照品储备液。精密吸取芦荟大黄素储备液6.4 mL、大黄酸储备液6 mL、大黄素储备液2 mL、大黄酚储备液4 mL、大黄素甲醚储备液1 mL,置于20 mL 容量瓶中,加甲醇至刻度,摇匀,配置成芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚浓度分别为0.048 2、0.067 5、0.409 0、0.122 0、0.399 5 mg/mL 的混合对照品溶液。
1.6 样品溶液制备 取本品适量,研细,取约0.3 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,称定重量,加热回流1 h,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过。精密量取续滤液5 mL,蒸干,加8%盐酸溶液15 mL,超声处理2 min,再加三氯甲烷10 mL,加热回流1 h,放冷,置分液漏斗中,用少量三氯甲烷洗涤容器,并入分液漏斗中,分取氯仿层,酸液再氯仿提取3 次,15 mL/次,合并氯仿层,蒸干。残渣加甲醇溶解,置10 mL 量瓶中,加甲醇至刻度摇匀,过0.45 μm 微孔滤膜滤过(10 μL 进样),取续滤液,即得。
2 结 果
2.1 HPLC 含量测定方法的建立
2.1.1 样品和混合对照品色谱图“1.4”色谱条件下混合对照品和样品色谱图见图1。
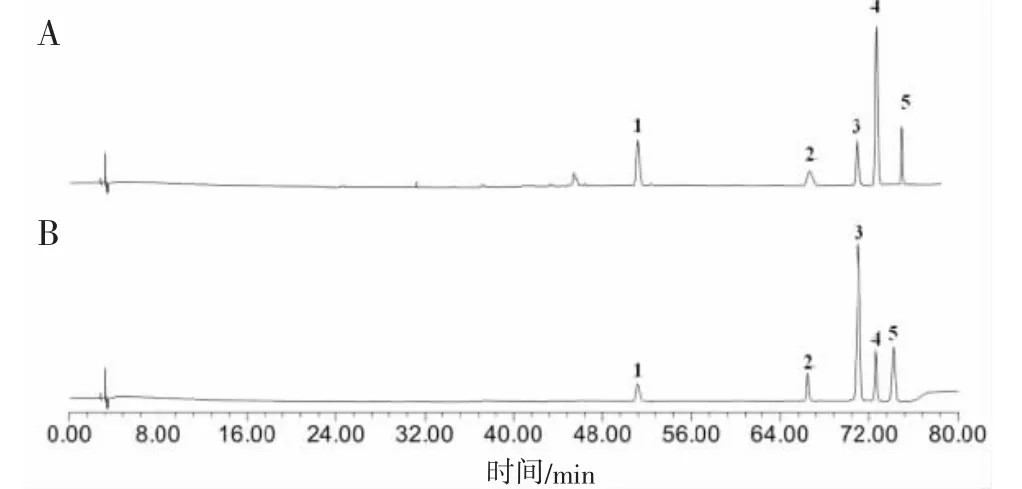
图1 样品(A)和混合对照品(B)色谱图
2.1.2 线性关系考察 精密吸取混合对照品溶液进样测定,计算5 个成分的回归方程及线性范围,见表1~2。
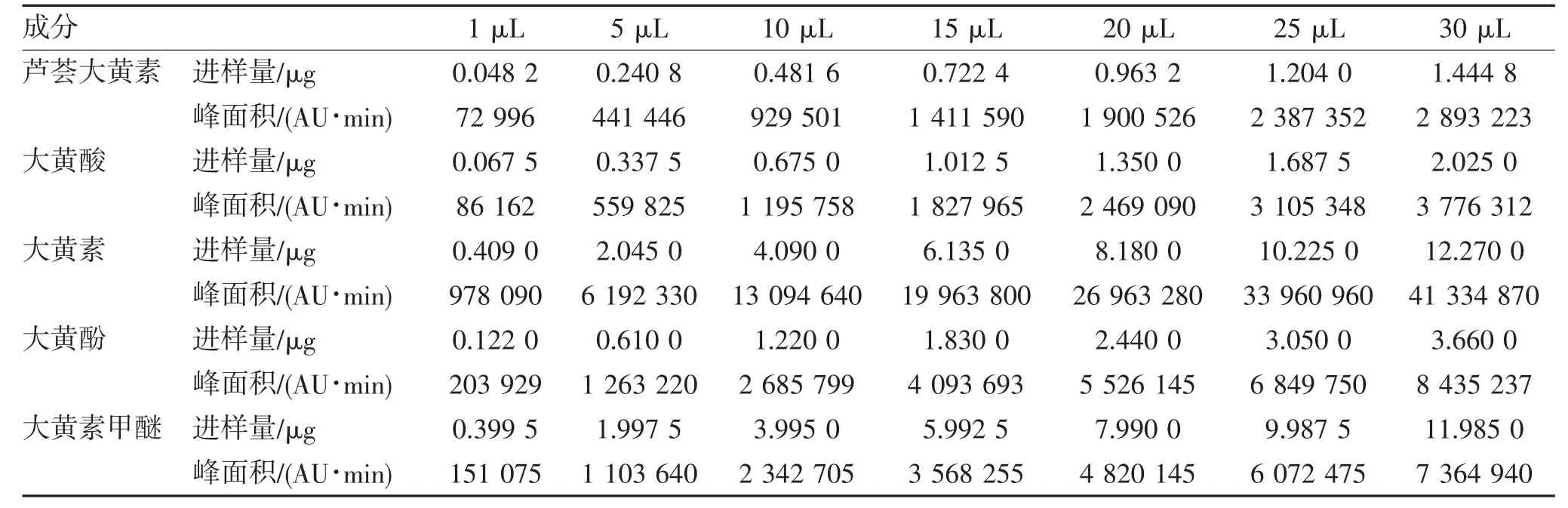
表1 5 个成分的进样量和峰面积
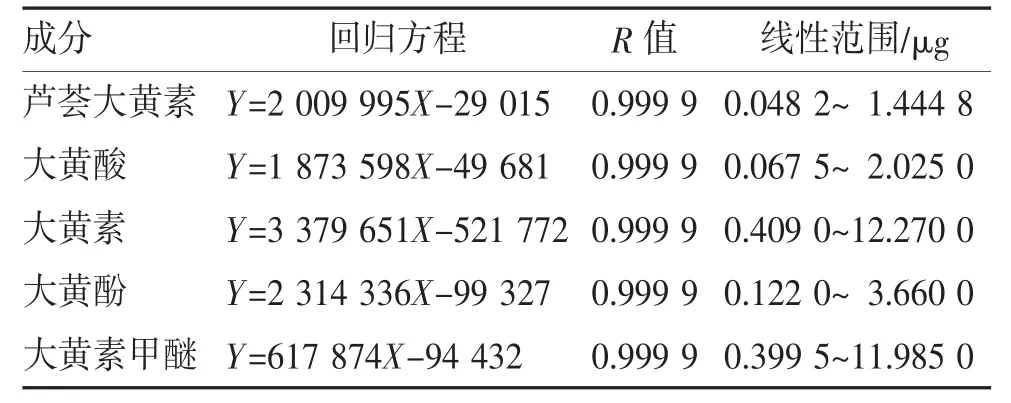
表2 5 个成分回归方程及线性范围
2.1.3 精密度试验 精密吸取混合对照品溶液10 μL连续进样测定,计算6 次结果的相对标椎偏差(relative standard deviation,RSD),结果表明仪器精密度良好,见表3。
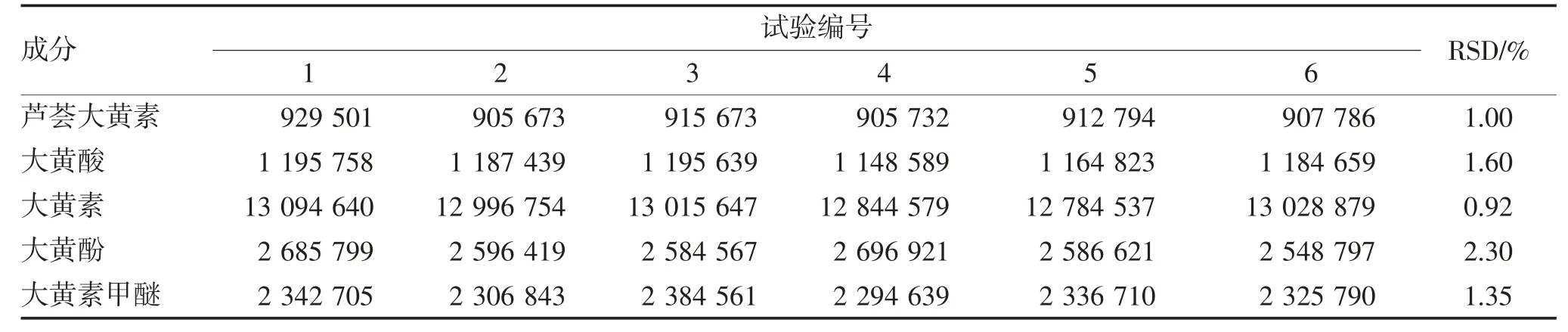
表3 精密度试验结果
2.1.4 稳定性试验 精密吸取样品溶液10 μL,于不同时间点进样测定,计算6 次结果的RSD,结果表明样品溶液在36 h 内均稳定,见表4。
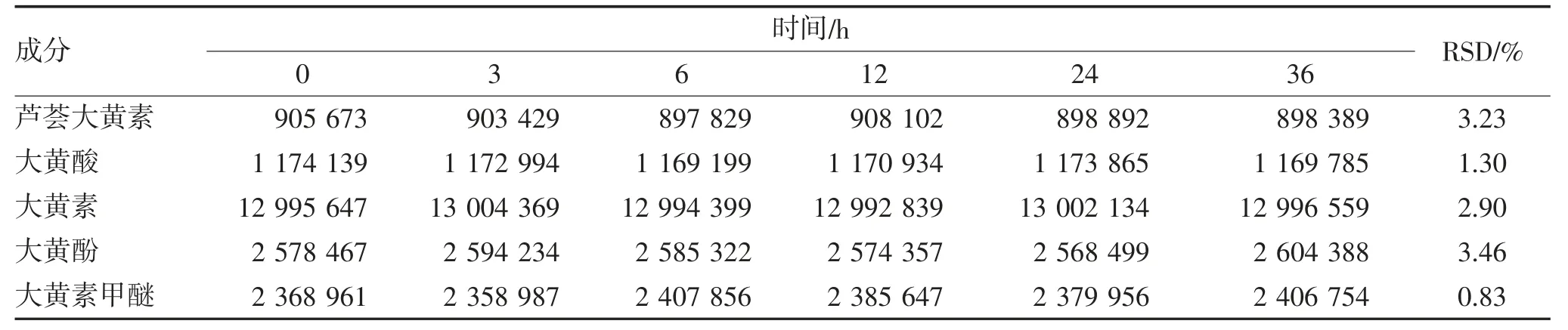
表4 稳定性试验结果
2.1.5 重复性试验 称取6 份九制大黄丸粉末,每份约0.3 g,制备成6 个样品溶液,各精密吸取10 μL进样测定,计算5 个蒽醌类成分含量测定结果的RSD,结果表明样品处理方法的重复性较好,见表5。
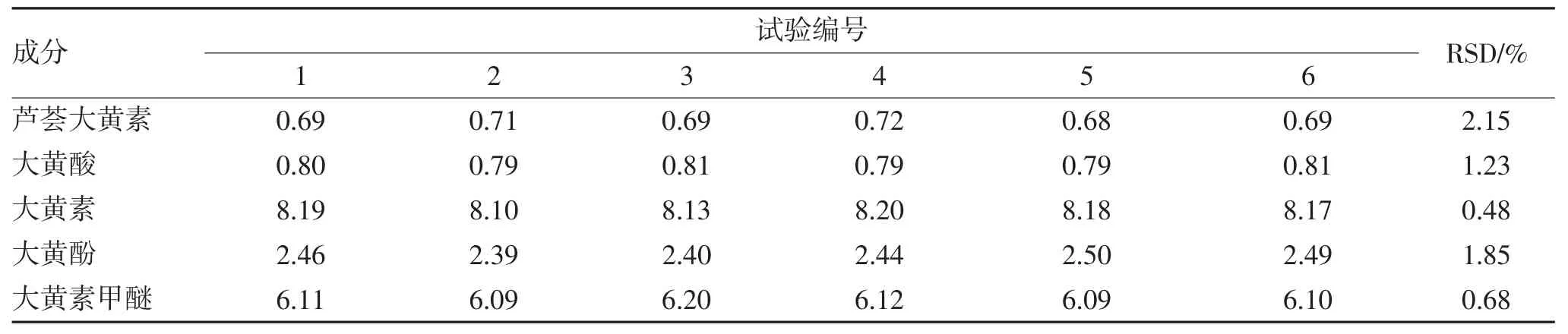
表5 重复试验结果
2.1.6 回收率试验 取已知含量的样品6 份,约0.15 g/份,精密称定,分别置25 mL 具塞锥形瓶中,各精密加入混合对照品溶液10 mL 左右,再各加甲醇12 mL 左右,按“1.6”方法制成样品溶液,测定并计算各成分的加样回收率及RSD,见表6。
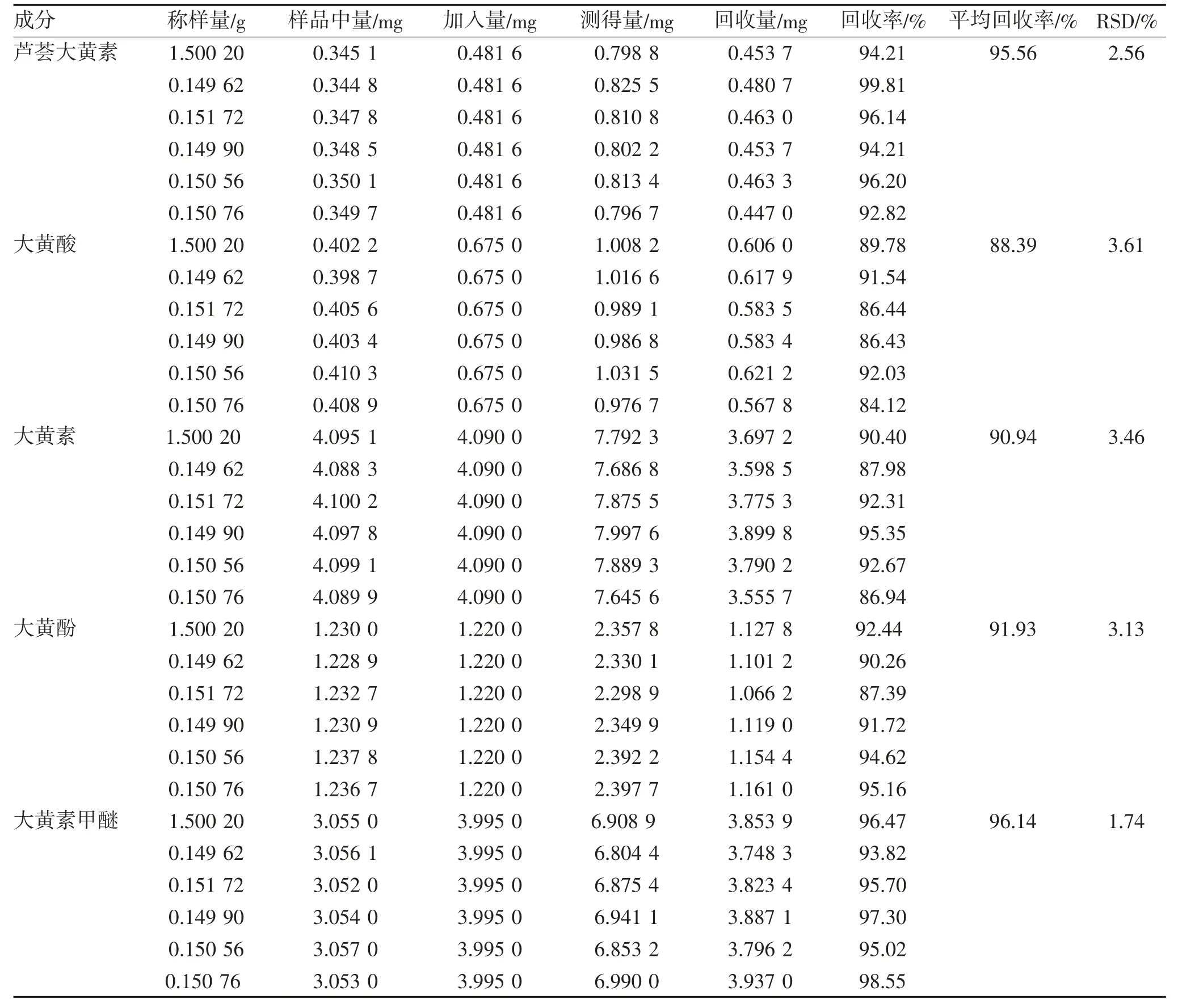
表6 加样回收率试验结果
2.2 校正因子的计算 以大黄素为内参物,取混合对照品溶液6 个进样体积折合成进样量(质量)计算所得的RCF,取平均值作为定量用fs/i,并考察了7 个质量点计算fs/i的RSD。RCF 计算公式[1]:fs/i=(Ws×Ai)/(Wi×As),其中As为内参物峰面积,Ws为内参物质量,Ai为某待测组分i 峰面积,Wi为某待测组分i 质量。计算待测组分芦荟大黄素(B)、大黄酸(C)、大黄酚(D)、大黄素甲醚(E)与内参物大黄素(A)的RCF。结果表明,4 个待测组分与内参物成分6 个质量点计算fs/i的RSD 均<5%,见表7。
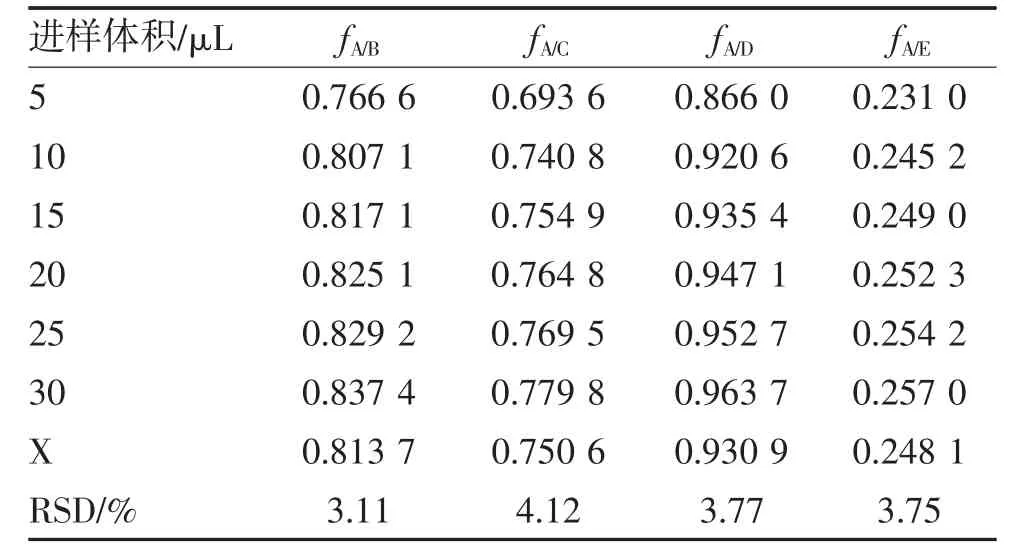
表7 4 个待测成分与大黄素的RCF
2.3 校正因子的重现性考察 精密吸取混合对照品溶液10 μL,进样分析,在两个实验室分别考察了Waters、Dionex 两种HPLC 系统和Symmetry C18(4.6 mm×250 mm,5 μm)、Aglilent TC-C18(4.6 mm×250 mm,5 μm)两种色谱柱对RCF 的影响,结果表明RSD 均<5%,表明在不同实验室、不同色谱系统和不同色谱柱下具有良好的重现性,见表8。
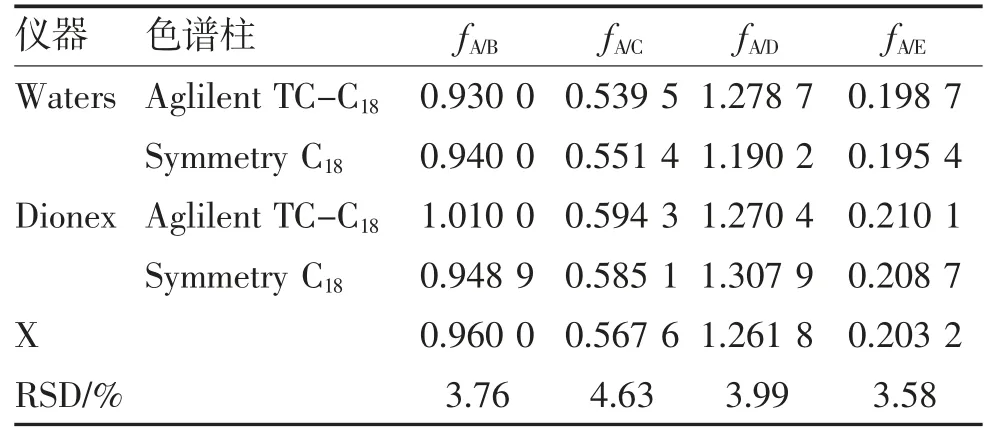
表8 不同仪器和色谱柱测定RCF
2.4 QAWS 与外标法测定结果的比较 分别精密吸取1~10 号样品溶液各10 μL,注入HPLC 仪测定。采用外标法(将各组样品中5 个蒽醌成分的峰面积代入“2.1.2”中5 个成分回归方程,计算出进样量,进而根据进样体积求样品中的量)测定5 个蒽醌成分的含量,再用建立的QAWS(根据“2.2”中RCF 计算公式)进行含量计算,并比较相对误差,结果表明,所有样品芦荟大黄素、大黄酚、大黄酸、大黄素甲醚这两种方法所得结果相对误差的绝对值基本<5%,提示建立的方法用于大黄素与其他4 种成分(芦荟大黄素、大黄酸、大黄酚、大黄素甲醚)之间进行含量测定具有较好的可信度,见表9。
由表9 可得本研究通过测定大黄素与其他4 个成分(芦荟大黄素、大黄酸、大黄酚、大黄素甲醚)之间的RCF 等方法最终确立的一测多评法具有良好的稳定性、可行性,可用于九制大黄丸中5 种成分的含量测定,为该中成药的质量控制提供依据。

表9 QAWS 与外标法测得的5 个蒽醌含量 mg/g
3 讨 论
3.1 内参物的选择 大黄素出峰顺序居中且与其他4 种成分分离度较高,对照品易获得,故选大黄素作为内参物[5]。3.2 色谱峰的定位 有学者[9]采用对照品建立线性方程,进行待测成分色谱峰定位,考察不同线性回归法在不同色谱柱上对待测组分色谱峰定位。本实验采用的是对照品色谱峰的比对定位。
3.3 QAWS 的优缺点 该方法可解决在针对药材质量控制方面对照品难以分离、单体不稳定及价格昂贵等问题,但是某些药材成分会因产地不同等因素而导致个别成分含量差异巨大,此时该方法适用面可能覆盖不到含量差异巨大的成分。
3.4 应用前景 本研究建立了QAWS,更加简洁、快速、经济、有效地实现了九制大黄丸中5 种蒽醌类成分的含量测定。《中国药典》2015 年版一部将一测多评法作为中药质量控制的重点推广技术并应用于丹参、生姜、黄连等药材,银杏叶提取物及制剂银杏叶片、银杏叶胶囊、银杏叶滴丸,咳特灵片、咳特灵胶囊等的质量控制。《美国药典》34 版、《欧洲药典》7.0 版中也分别有25 个和9 个天然药物及制剂应用该方法进行质量控制。该方法极大地缩减了药学工作中所需花费的人力物力,并可有效地对药物进行质量控制[10]。
