同构MOFs复合氧化石墨烯电极材料构筑高性能超级电容器
2021-11-18籍文娟王国娇孙秀玲付云龙
籍文娟 王 丹 王国娇 孙秀玲 付云龙
(山西师范大学化学与材料科学学院,磁性分子与磁信息材料重点实验室,太原 030006)
0 引 言
超级电容器(SCs)是一种新型、清洁、高效的电能转换和储存设备[1⁃3],具有小巧轻便、功率密度高[4⁃5]、充电速度快[6⁃7]、循环寿命长[8⁃9]、成本低[10]的优点。近年来,以金属有机框架材料(MOFs)作为前驱体制备多种电极材料的研究已经备受关注。MOFs具有大的比表面积[11⁃13]、可调的孔隙结构、一体化的氧化还原金属中心,这使其表现出很好的电化学活性,成为电化学储能领域的明星材料[14⁃19]。但是,MOFs本身具有导电性和稳定性较差的缺点,限制了其在超级电容器方面的应用[20⁃21]。
目前,将MOFs进行碳化或与其他材料复合是优化其电化学性能的主要方法。将MOFs碳化得到的金属氧化物或多孔碳直接作为电极材料时循环稳定性较好,但比电容较低,例如基于HKUST⁃1[22]、MOF⁃505[23]和 Al⁃PCP[24]为前驱体的碳化材料中,以Al⁃PCP为前驱体的碳材料在100 mA·g-1电流密度下比电容为 232.8 F·g-1[24]。聚苯胺[25]、石墨烯[26]与MOFs复合也可优化MOFs的导电性,而氧化石墨烯(GO)作为一种很好的候选复合材料具有以下优点:首先,GO的2D薄层两侧存在羟基、羧基、羰基、环氧等含氧官能团,可以吸引金属离子,更容易实现与MOFs的复合[27⁃28]。其次,GO能增加MOFs内的分散力,影响 MOFs的结晶[29⁃30],进而调控 MOFs的结构、形态和晶体的尺寸[31⁃32]。最后,GO与MOFs存在协同作用,复合后的材料同时表现出双电层和赝电容特性,增大离子扩散速率,减小电阻,使复合材料表现出比复合前更好的电化学性能[33⁃34]。
首先,我们选择具有较好稳定性的高核和高连接的Zn⁃MOF为结构模型[35],引入具有氧化还原活性的Co和Ni金属,合成系列同构的Co⁃MOF、Ni⁃MOF,进而调控电极材料的电化学性能。其次,由于 X⁃MOF X6O(TATB)4(H+)2·(H2O)8·(DMF)2,X=Zn、Co、Ni;H3TATB=4,4′,4″⁃s⁃triazine⁃2,4,6⁃triyl⁃tribenzoic acid;DMF=N,N⁃二甲基甲酰胺)本身的导电性较差,因此选用高导电性的GO进一步与其复合,合成系列X⁃MOF@GO复合材料,并系统地研究其电化学性能。
因此,我们在水热条件下以GO为结构导向剂,调控系列硝酸盐((X(NO3)2,X=Zn、Co、Ni)和配体(H3TATB)的自组装过程,合成文献[35]报道的同构X⁃MOF,其中 Zn⁃MOF结构如下:4个三核 Zn簇(Zn3(COO)6)通过μ4⁃O 相连接,形成 12核金属簇{[Zn3(COO)6)4]O}4(图 1a)。6 个四核 Zn 簇(Zn4O)(顶点)与线性三核簇(边)连接而成八面体笼;以八面体面上的4个四核簇为顶点,首尾连接成内四面体笼(八面体笼嵌套四面体笼结构)(图1b);八面体笼之间采取共顶点的方式拓展成具有堆积孔的三维结构(图1c)。进而利用表面含有羟基、羧基、羰基、环氧等含氧官能团的GO与金属离子配位,通过抑制结晶过程中的聚集,调控形成尺寸更小的MOFs颗粒,进而诱导形成系列尺寸更小的X⁃MOF@GO材料(图2),并研究了其作为超级电容器的电化学性能。
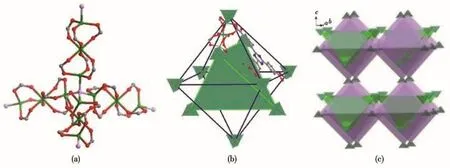
图1 Zn⁃MOF的晶体结构图[35]Fig.1 Crystal structure of Zn⁃MOF[35]
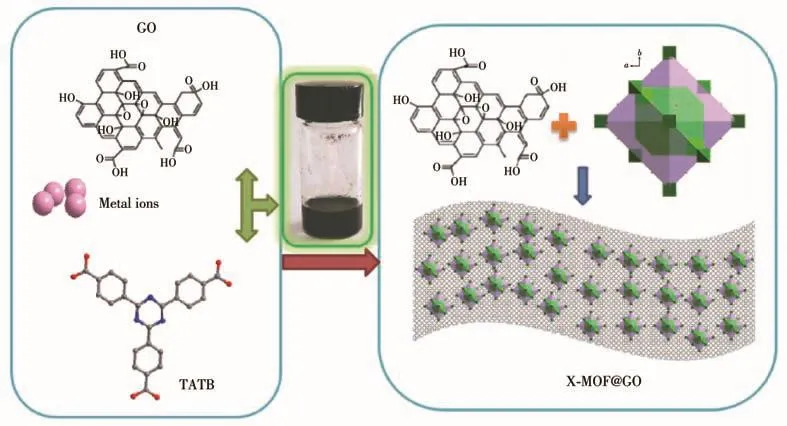
图2 X⁃MOF@GO(X=Zn、Co、Ni)的合成方案Fig.2 Synthetic scheme of X⁃MOF@GO(X=Zn,Co,Ni)
1 实验部分
1.1 试剂
六水合氯化钴(CoCl2·6H2O)、四水合醋酸锌(Zn(Ac)2·4H2O)、六水合氯化镍(NiCl2·6H2O)均购于阿拉丁生化科技股份有限公司;乙炔黑购于天津晶林新材料科技有限公司;聚偏氟乙烯(PVDF)分散液购于太原迎泽力之源电池材料有限公司;N,N⁃二甲基乙酰胺(DMA)和N⁃甲基吡咯烷酮(NMP)购于梯希爱(上海)化成工业发展有限公司;GO水溶液(5 mg·mL-1)购于苏州恒球科技有限公司;H3TATB购于济南恒化科技有限公司。
1.2 表征方法
采用UltimaⅣ⁃185(日本理学公司)X射线粉末衍射仪(PXRD)分析样品物相组成,MoKα辐射(λ=0.071 073 nm),电压40 kV,电流40 mA,扫描范围5°~50°,扫速为 20(°)·min-1。采用 VARIAN 660⁃IR型傅里叶红外光谱仪测试样品的IR光谱,测试范围4 000~400 cm-1。采用GA/DSC 1型热分析仪测试样品的TG曲线,从室温升至800℃,空气氛围,升温速率5℃·min-1。采用德国卡尔蔡司(Carl Zeiss)公司的JSM⁃7500F型场发射扫描电子显微镜(SEM)对样品表面微观形貌进行分析,加速电压10 kV。通过Rigaku MLtimaⅣ⁃185 X射线光电子能谱仪(XPS)获得其表面的元素种类、元素化合价及其含量。
1.3 系列X-MOF(X=Zn、Co、Ni)的合成
依据文献[35]并优化条件以合成X⁃MOF,具体如下:将0.035 g配体(H3TATB)、0.015 g Zn(Ac)2·4H2O、3.0 mL DMA、1 mL去离子水在常温下超声混合1 h后转移到密闭的20 mL无色透明玻璃小瓶中,在100℃恒温烘箱中加热反应72 h,待冷却至室温,用DMA冲洗并干燥后得到乳白色块状晶体(Zn⁃MOF)。Ni⁃MOF与Co⁃MOF的合成方法同上,只需将Zn(Ac)2·4H2O分别用NiCl2·6H2O、CoCl2·6H2O 替换,制备出与Zn⁃MOF结构相同的Ni⁃MOF和Co⁃MOF粉末。
1.4 系列X-MOF@GO(X=Zn、Co、Ni)的合成
将 0.035 g配体(H3TATB)、0.015 g Zn(Ac)2·4H2O加入到1 mL GO和3 mL DMA的混合液中,在100℃下恒温反应48 h,最终制得Zn⁃MOF@1.0GO。采用同样的方法分别获得Ni⁃MOF@1.0GO和Co⁃MOF@1.0GO。同时,在制备Ni⁃MOF@GO时,改变GO用量(1.5和2.0 mL),分别获得Ni⁃MOF@1.5GO、Ni⁃MOF@2.0GO。
1.5 电极制备与电化学性能测试
三电极工作电极的制备:按质量比8∶1∶1称取X⁃MOF@GO、乙炔黑、PVDF并分散到NMP溶剂中,然后涂到预先处理过的泡沫镍上(约1 cm2),于60℃下真空干燥12 h后用粉末压片机(JSP⁃12)在10 MPa下压片。
二电极正、负电极片的制备:根据如下公式[13]计算得出制备正极所需的活性材料的质量,其中,m+和m-、C+和C-、ΔV+和ΔV-分别为正、负极的质量、比电容、电压范围。按上述方法制备电极片。
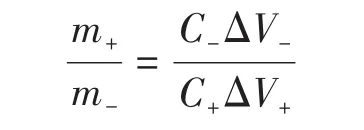
电化学性能测试:以1 mol·L-1KOH溶液为电解液,铂片(1 cm×1 cm)为对电极,饱和甘汞电极为参比电极,并在室温下对其进行循环伏安(CV)、恒电流充放电(GCD)、交流阻抗(EIS)等测试。
2 结果与讨论
2.1 IR分析
对X⁃MOF、X⁃MOF@1.0GO(X=Zn、Co、Ni)和 GO进行IR谱图测试(图3)。GO的IR谱图中位于3 438 cm-1处强特征峰为—OH的伸缩振动峰,1 732 cm-1为羧基中的C=O伸缩振动峰,1 071 cm-1为羧基内的C—O伸缩振动峰,1 224 cm-1为C—O—C键的伸缩振动峰,1 634 cm-1处为GO吸附水的弯曲振动峰。X⁃MOF与X⁃MOF@1.0GO(X=Zn、Co、Ni)的IR谱图类似,其中810、776、692 cm-1处为苯环上间位取代的吸收峰,1 015 cm-1处为配体中羧基内的C—O伸缩振动峰,1 014 cm-1处为C—C键的特征峰,1 358和1 403 cm-1处的吸收峰为三嗪环上的C—N键的伸缩振动峰,1 660 cm-1处为羧基中的C=O键的特征峰,1 568和1 502 cm-1处为C=C键的特征峰。

图3 X⁃MOF、X⁃MOF@1.0GO(X=Zn、Co、Ni)及GO的IR谱图Fig.3 IR spectra of X⁃MOF,X⁃MOF@1.0GO(X=Zn,Co,Ni)and GO
2.2 TG分析
图4 是样品的TG曲线。由图可知,各样品的下降趋势相似,在25~200℃范围内,质量的损失主要来自样品中溶剂分子的脱去。样品在450℃时框架依然稳定,表明其具有良好的热稳定性。
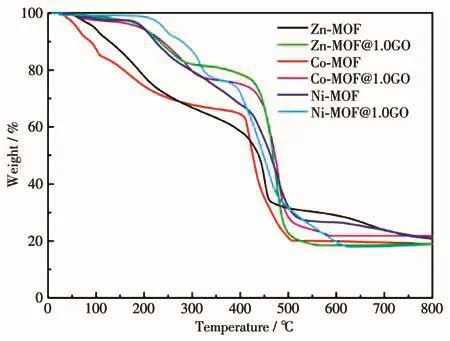
图4 X⁃MOF和X⁃MOF@1.0GO(X=Zn、Co、Ni)的TG曲线Fig.4 TG curves of X⁃MOF and X⁃MOF@1.0GO(X=Zn,Co,Ni)
2.3 PXRD表征
从图5中可以看出,制备的X⁃MOF和X⁃MOF@GO(X=Zn、Co、Ni)的PXRD峰位置与模拟衍射峰[35]匹配良好,表明复合前后的粉末纯度较高,并且有着相似的框架结构,说明GO的引入未影响晶体的框架结构。
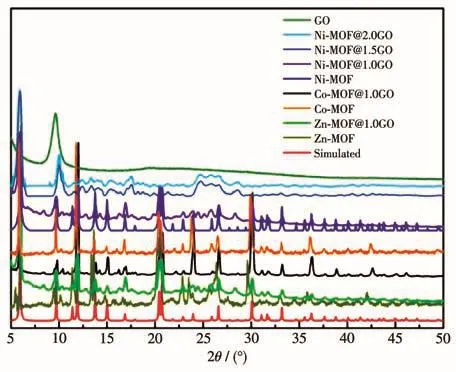
图5 X⁃MOF、X⁃MOF@GO(X=Zn、Co、Ni)和GO的PXRD图Fig.5 PXRD patterns of X⁃MOF,X⁃MOF@GO(X=Zn,Co,Ni)and GO
2.4 XPS分析
图6 为样品的XPS谱图。在1 022.1和1 045.2 eV 处是 Zn2p3/2和 Zn2p1/2的特征峰(图 6a);781.6 和797.8 eV处是Co2p3/2和Co2p1/2的特征峰(图6c);856.1和874.1 eV处是Ni2p3/2和Ni2p1/2的特征峰(图6e)。由图可知,复合GO前后Zn2p、Co2p、Ni2p的峰位几乎无变化,表明GO并未影响MOF中金属离子的价态。另外,在C1sXPS谱图中,复合前样品均出现288.7 eV(C=O)、286.6 eV(C—O)、284.8 eV(C=C/C—C)的特征峰。但复合GO后还出现了对应于GO中羧基(O—C=O)的特征峰,峰位分别为291.7 eV(Zn⁃MOF@1.0GO)、291.6 eV(Co⁃MOF@1.0GO)、290.8 eV(Ni⁃MOF@1.0GO)[15⁃16],这说明 X⁃MOF(X=Zn、Co、Ni)与GO成功复合。
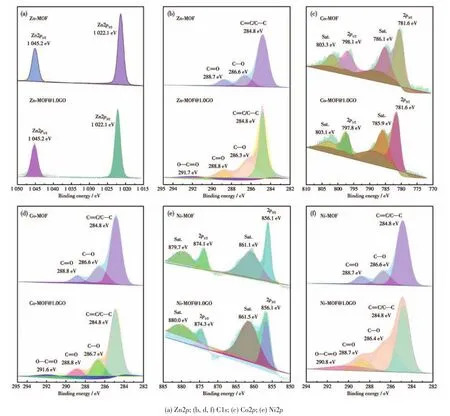
图6 X⁃MOF及X⁃MOF@1.0GO(X=Zn、Co、Ni)的XPS谱图Fig.6 XPS spectra of X⁃MOF and X⁃MOF@1.0GO(X=Zn,Co,Ni)
2.5 SEM表征
从图7a中可看出,复合前GO为片状结构,复合后分散为更薄的层状结构(图7b)。从图S1a(Support⁃ing information)可知,Zn⁃MOF具有八面体的单晶结构,其尺寸为 40~60 μm;如图 7c所示,GO 与Zn⁃MOF复合后,其八面体的表面变得比较粗糙且可看到GO的褶皱,这表明GO与Zn⁃MOF复合成功。由图S1b可知,Co⁃MOF也具有八面体的形貌,其尺寸约为250 μm。图7d为Co⁃MOFs@1.0GO的 SEM图。图S1c表明Ni⁃MOF为类立方体状,其尺寸为30~60 μm。Ni⁃MOF、Ni⁃MOF@1.0GO的晶体形貌不同可能是晶体生长的机制不同所致[36⁃37]。当GO的加入量为1 mL时,Ni⁃MOF晶体由类立方体变为片层状且附着在片状GO上(图7e),GO的量相比Ni⁃MOFs晶体较少;随着GO用量变为1.5 mL,Ni⁃MOF块状表面上有褶皱的GO(图7f),这增强了其导电性,有利于电子和离子的传输。但过量GO(2 mL)加入时样品发生团聚(图S1d),阻碍了离子和电子的传输。从Ni⁃MOF@1.5GO的TEM(图S1e)中可以看出,Ni⁃MOF特定的框架结构和GO表面的官能团相互作用使其生长在GO的表面。
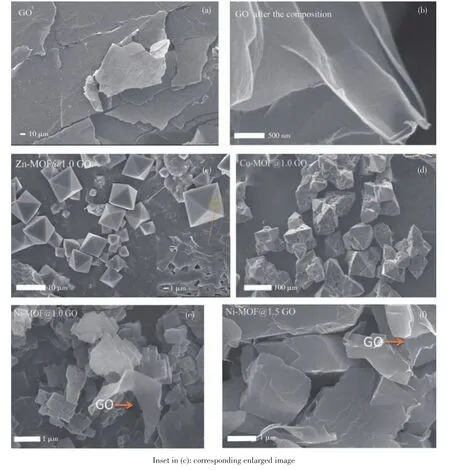
图7 (a、b)GO和(c~f)X⁃MOF@GO(X=Zn、Co、Ni)的SEM图Fig.7 SEM images of(a,b)GO and(c⁃f)X⁃MOF@GO(X=Zn,Co,Ni)
2.6 三电极体系中的电化学测试
2.6.1 CV分析
X⁃MOF 及 X⁃MOF@1.0GO、Ni⁃MOF@1.5GO、Ni⁃MOF@2.0GO的CV曲线如图S2a~S2c和8a~8e所示,电势窗口为-0.1~0.8 V。由图可知,扫描速率逐渐增大(5、10、20、50 mV·s-1)时,样品的CV曲线的面积逐渐变大,阳极峰和阴极峰电位均发生偏移,氧化还原峰也逐渐消失,呈现出典型的赝电容特征,而非双电层电容的特征。根据Randles⁃Sevcik方程中氧化还原峰的峰值电流(ip)与扫描速率(v)之间的关系,进一步确定电荷存储过程中电化学反应的机理。当ip与v成正比,则说明样品表面发生氧化还原反应;而当ip与v1/2成正比时,则说明反应中存在固相扩散。
ip/v1/2=(2.69×105)n3/2AD1/2c0
其中,A为电极面积(cm2),D为扩散系数(cm2·s-1),c0为反应物的浓度(mol·cm-3),n为氧化还原反应中电子转移的个数。
如图8f所示,从X⁃MOFs的ip与v1/2关系图可看出,随着扫描速率的增加,电流增大,表明扩散过程中电子传递速率快,由线性拟合函数关系图,可以看出电极的氧化还原过程和固相扩散控制。另外,斜率越大,扩散系数越大,与GO复合后的X⁃MOF@GO的斜率均大于未复合前的样品X⁃MOF,说明GO的掺入更有利于氧化还原过程中电子在固相中扩散传输。其中Co⁃MOF@1.0GO的斜率最大,说明其扩散系数最大,复合后更有利于氧化还原过程的进行和固相扩散。但是,离子扩散到MOF孔道的阻力也影响其扩散过程,导致Ni⁃MOF@1.5GO的扩散系 数 略 小 于 Co⁃MOF@1.0GO[20⁃21]。 图 中 Ni⁃MOF@2.0GO的线性相关性较差,说明GO量增多不利于氧化还原和固相扩散的进行[20⁃21]。
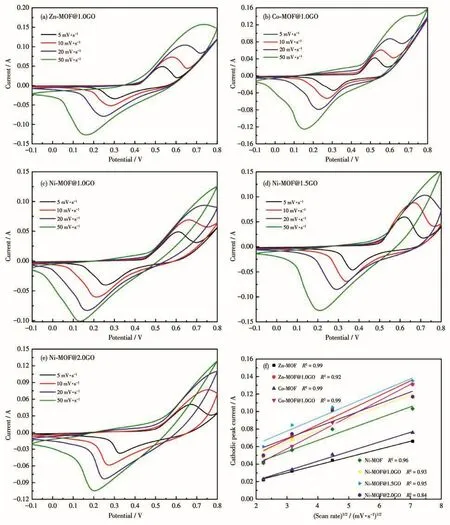
图8 (a~e)X⁃MOF@GO(X=Zn、Co、Ni)在不同扫描速率下的CV图;(f)ipvs v1/2的关系Fig.8 (a⁃e)CV curves of X⁃MOF@GO(X=Zn,Co,Ni)at various scanning rates;(f)Relationship of ipvs v1/2
2.6.2 GCD测试分析
GO、X⁃MOF和X⁃MOF@GO电极的GCD曲线如图S3、图9所示。由图可知,在不同电流密度(0.5、1、2、3 A·g-1)下,电极的充放电曲线对称性较好,进一步表明X⁃MOF和X⁃MOF@GO电极材料的赝电容特性,即通过发生氧化还原反应来存储电荷。复合GO后,充放电时间都明显增加,根据以下公式计算比电容:
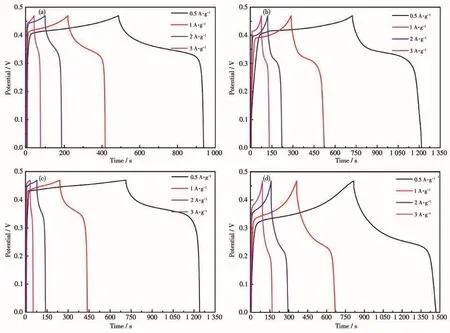
图9 (a)Zn⁃MOF@1.0GO、(b)Co⁃MOF@1.0GO、(c)Ni⁃MOF@1.0GO、(d)Ni⁃MOF@1.5GO在不同电流密度下的GCD曲线Fig.9 GCD curves of(a)Zn⁃MOF@1.0GO,(b)Co⁃MOF@1.0GO,(c)Ni⁃MOF@1.0GO,and(d)Ni⁃MOF@1.5GO at different current densities
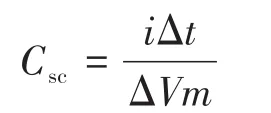
其中,i(A)、Δt(s)、m(g)分别代表放电电流、放电时间、负载在泡沫镍上的活性材料质量。各电极相应的比电容见表S1。
为进一步考察GO用量对比电容的影响,选择比电容较高的Ni⁃MOF进行GO用量的调控研究,结果如图9所示。由图可知,比电容的大小与GO的复合量并不成正比。其中,Ni⁃MOF@1.5GO相对Ni⁃MOF@1.0GO和Ni⁃MOF@2.0GO表现出更大的比电容,这可能由于较少的GO会降低电子转移率,而过量的GO使得晶体团聚,导致离子迁移受阻,从而影响比电容的大小。同时,复合GO的材料比电容明显增大。
由图10可以看出,在0.5 A·g-1时,X⁃MOFs@GO电极材料的比电容都相对其X⁃MOF电极有所提高,其中,Zn⁃MOF@1.0GO 的比电容增加 162.6 F·g-1,Co⁃MOF@1.0GO的比电容增加 194.1 F·g-1,Ni⁃MOF@1.0GO的比电容增加141.6 F·g-1,均高于文献报道的部分材料[20⁃21,38⁃39]。其优异的电化学性能可能是X⁃MOF结构与GO中排列密集的原子之间的协同效应,使得复合GO后X⁃MOFs的表面分散力较大,影响了晶体的大小,使其暴露出更多的法拉第反应的金属活性位点,确保了足够多的能量存储。
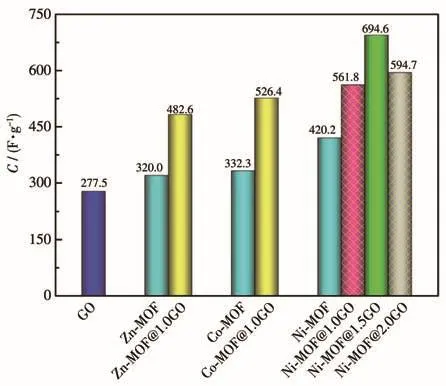
图10 GO、X⁃MOF及X⁃MOF@GO(X=Zn、Co、Ni)在0.5 A·g-1下的比电容值Fig.10 Specific capacitance values for GO,X⁃MOF,and X⁃MOF@GO(X=Zn,Co,Ni)at 0.5 A·g-1
2.6.3 交流阻抗分析
为了进一步评估这些材料的动力学机理,我们进行了EIS测试。如图11所示,X⁃MOF@GO(X=Zn、Co、Ni)的Rct(法拉第电阻)比X⁃MOF 的Rct明显较小,说明复合后的电极具有更快的电荷转移能力,电解液中的OH-离子能够更快地扩散到电极表面。

图11 X⁃MOF及X⁃MOF@1.0GO(X=Zn、Co、Ni)的EIS谱图Fig.11 EIS spectra of X⁃MOF and X⁃MOF@1.0GO(X=Zn,Co,Ni)
2.6.4 循环寿命分析
循环稳定性是评价电极材料实用性的重要参数。根据图12可知,利用蓝电测试系统(型号:CT2001A)在3.5 A·g-1电流密度下,连续循环1 000次后,Zn⁃MOF、Zn⁃MOF@1.0GO、Co⁃MOF、Co⁃MOF@1.0GO、Ni⁃MOF、Ni⁃MOF@1.0GO的比电容保持率分别为初始比电容的56.8%、85.6%、52.9%、82.5%、65.3%、81.2%。可见GO的复合可提高X⁃MOFs的循环稳定性。
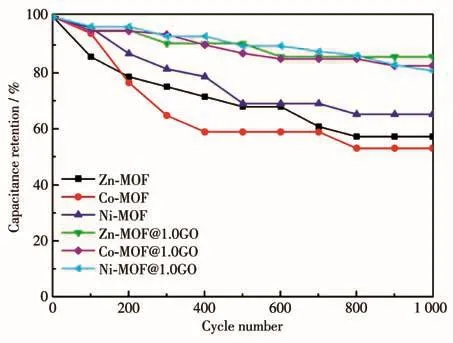
图12 X⁃MOFs和X⁃MOFs@1.0GO的循环稳定性Fig.12 Cyclic stability of X⁃MOFs@1.0GO
2.6.5 二电极CV测试分析
在三电极测试中,Ni⁃MOF@1.5GO表现出了良好的电化学性能,因此我们只构建了非对称超级电容器Ni⁃MOF@1.5GO//AC来评估其实际应用性。图13a是电极材料的CV曲线。图13b是组装的非对称超级电容器Ni⁃MOF@1.5GO//AC的CV曲线。很明显,随着扫速的增加(5~50 mV·s-1),CV曲线围成的矩形面积增大,但其形状没有明显的畸变,显示了典型的非对称超级电容器的快速充放电行为。
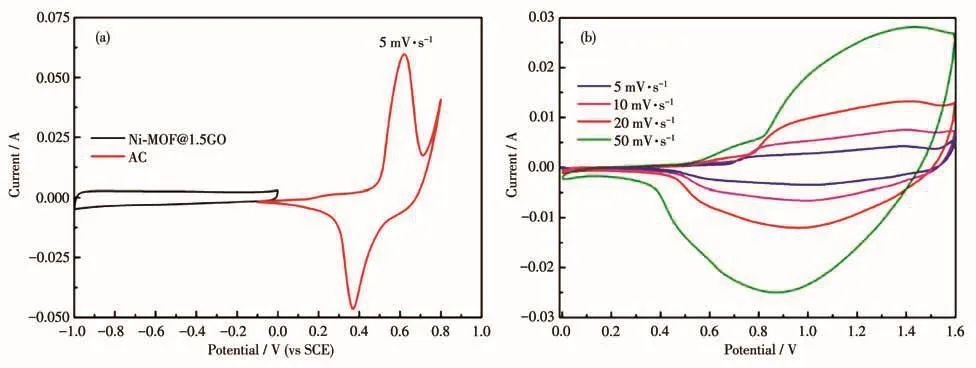
图13 (a)电极在5 mV·s-1时的CV曲线;(b)Ni⁃MOF@1.5GO//AC在不同扫速下的CV曲线Fig.13 (a)CV curves of electrodes at 5 mV·s-1;(b)CV curves of Ni⁃MOF@1.5GO//AC at different scanning rates
2.6.6 二电极的GCD测试分析
根据活化的Ni⁃MOF@1.5GO//AC非对称超级电容器的GCD曲线可知(图14),其最大电位可达1.5 V。此外,电流密度为1、2、3和5 A·g-1时比电容分别为49.2、42.9、37.1和30.0 F·g-1。可见,随着电流密度的增加,非对称超级电容器的比电容减小。这是因为电流密度较大时,电解液中离子迁移过快,导致电解液离子与金属活性位点接触不充分,因而,高电流密度时不利于电荷和离子传输,比电容相对较小。
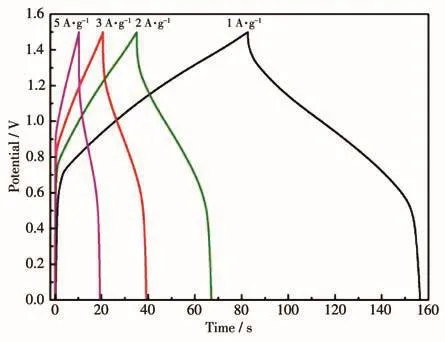
图14 Ni⁃MOF@1.5GO//AC在不同电流密度下的GCD曲线Fig.14 GCD curves of Ni⁃MOF@1.5GO//AC at different current densities
2.6.7 二电极循环稳定性测试分析
Ni⁃MOF@1.5GO//AC 在 1 A·g-1下 经 过 连 续3 000次充放电测试后,比电容量保持率约70.0%(图15),说明这种复合电极材料具有较好的循环稳定性。对循环后的电极材料进行形貌表征,如图15内插图所示,可知其仍为类立方体状的纳米颗粒,说明循环后样品的形貌基本没有发生变化。
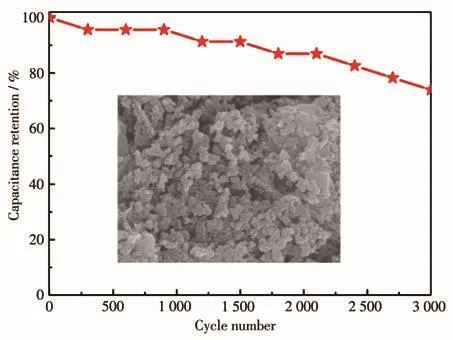
图15 Ni⁃MOF@1.5GO//AC的循环稳定性和SEM图(插图)Fig.15 Cycling stability and SEM image(Inset)of Ni⁃MOF@1.5GO//AC
2.6.8 功率密度和能量密度的计算
对于非对称超级电容器Ni⁃MOF@1.5GO//AC的性能评估还需结合其能量密度(E)和功率密度(P),计算公式如下:

其中,Δt(s)表示放电时间。进一步将E和P转换为Ragone图,如图S4所示。在电池电压为1.5 V时,Ni⁃MOF@1.5GO//AC 在P=754.3 W·kg-1时 输 出E=15.4 Wh·kg-1;在P=3.8 kW·kg-1时,输出E=9.4 Wh·kg-1。
3 结论
选择不同金属中心的同构MOFs并与GO复合制备系列X⁃MOF@GO材料。这3种复合材料的比电容相比原MOFs均有提高。在3.5 A·g-1的电流密度下循环1 000次后,Ni⁃MOF@1.0GO比电容保持率约81.2%。将Ni⁃MOF@1.5GO组装成非对称超级电容器 Ni⁃MOF@1.5GO//AC后,其在 1、2、3、5 A·g-1时的比电容分别为49.2、42.9、37.1、30.0 F·g-1,在功率密度为754.3 W·kg-1时,能量密度为 15.4 Wh·kg-1。本研究为MOF与GO的复合材料在超级电容器方面的应用提供了新的研究思路。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
杂志排行

无机化学学报的其它文章
- Heterometallic Ln(Na)-MOFs(Ln=Tb,Dy,Ho):Crystal Structures,Luminescent Sensing for Acetaldehyde,Fe3+,Cr2O72-,and Electrochemical Sensing for Catechol
- 自支撑多孔碳/硒复合柔性电极的制备及其电化学性能
- 松针状NiCo2O4@碳布复合材料在锂硫电池中的应用
- 1,4-二氮杂二环[2.2.2]辛烷-氰基合钴(Ⅲ)三维框架氢键型晶体的合成、相变及介电性质
- Ag@硅氧倍半聚合物的合成及其对对硝基苯酚的催化还原性能
- 介孔调控Co9S8/Ni3S2复合电极材料及电催化析氢性能