MGISEQ-2000、HiSeq 2000 与NovaSeq 6000平台全基因组重测序数据的比较分析
2021-11-18李伟宁唐中林刘剑锋孙飞舟
李伟宁,刘 刚,周 荣,唐中林,刘剑锋*,孙飞舟*
(1.中国农业大学动物科学技术学院,北京 100193;2.全国畜牧总站畜禽遗传资源保存利用中心,北京 100193;3.中国农业科学院北京畜牧兽医研究所,北京 100193;4.中国农业科学院农业基因组研究所,广东深圳 518120)
全基因组重测序(以下简称“重测序”)被广泛用于变异检测[1]、遗传成分鉴定[2]和多态性分析[3]等研究,现已成为疾病预测及诊断[4]、动植物分子育种[5-6]等领域最常用的分析方法之一。目前最常用的二代测序平台是Illumina(美国)测序平台,其在基因测序市场所占份额近75%。HiSeq 2000 是Illumina 在2010 年发布的一款测序仪,其将人类基因组测序费用降至1 万美元以下,大量生物公司和科研机构均采购了该测序仪,目前它仍是市场上的主流测序仪。NovaSeq 6000是Illumina 在2017 年发布的一款被誉为里程碑式产品的测序仪,可以搭配4 种不同的流动槽灵活地开展不同通量要求的测序任务,有望将基因组的测序费用进一步降至100 美元。国内的测序平台研发仍在起步阶段,2014 年6 月深圳华大智造科技股份有限公司(简称“华大智造”,英文名称为MGI)推出了BGISEQ-1000 和BGISEQ-100 2 个二代测序平台,是国家食品药品监督管理总局首次批准注册的第二代基因测序诊断产品,随后几年华大智造也陆续发布了多款适用于不同场景的二代测序仪。MGISEQ-2000[7]是华大智造在2017 年9 月推出的一款产品,该平台采用了该公司自主研发的CoolMPS[8]高通量测序试剂和DNA 纳米球测序技术,可在测序过程中实现高准确性、低重复序列率和低标签跳跃率,其凭借卓越的性能表现及超高性价比在众多测序平台中脱颖而出。
相关研究比较了Illumina 与华大智造二代测序平台的性能表现。Huang 等[9]研究发现BGISEQ-500的比对质量要好于HiSeq 2500,且二者的SNP 检测准确性均在99% 以上。Korostin 等[10]对比分析了MGISEQ-2000 和HiSeq 2500 的测序数据,发现在原始数据质量、变异检测方面二者表现相似,但MGISEQ-2000 的比对质量要优于HiSeq 2500。但上述研究的样本量较少,平台也较为单一。MGISEQ-2000和NovaSeq 6000 作为华大智造和Illumina 同期发布的2 款测序仪在性能表现上是否有明显差异,NovaSeq 6000 与Illumina 早期发布的HiSeq 2000 相比性能又是否有明显提升,还需要通过实际数据进一步验证。本研究基于MGISEQ-2000、HiSeq 2000 和NovaSeq 6000 3个平台对同一批样品进行重测序分析,比较不同平台的性能表现和测序稳定性,为研究者在选择不同测序平台时提供参考。
1 材料与方法
1.1 实验动物及样品 本研究实验动物为9 头公猪(4头马身猪、5 头大河猪),采集所有实验动物的耳组织,浸没于75%的酒精,置于-80℃冰箱保存备用。采用天根生物科技(北京)有限公司生产的组织基因组DNA提取试剂盒,严格按照产品说明书提取实验样本耳组织的全基因组DNA,采用酶标仪测定待实验样本的基因组DNA 浓度与纯度,检测合格后进行后续文库构建。
1.2 全基因组重测序及SNP 芯片分型 将9 头公猪的DNA 样本各自均分为3 份,按照MGISEQ-2000、HiSeq 2000 和NovaSeq 6000 3 个测序平台的标准建库流程分别对每份样本进行建库,在3 个平台上均采用paired-end、PE150(即双端150 bp 读长)对样本进行全基因组重测序,测序深度大于20X。全基因组SNP芯片在基因分型上具有很高的准确性[11],通常作为评价测序数据SNP 检测准确性的金标准[12-13]。本研究用猪50K 芯片(参考基因组版本为Sscrofa10.2)对所有样本进行了SNP 分型,作为评价重测序数据SNP 检测准确性的依据。
1.3 序列比对及变异检测 本研究分析步骤及所用软件如图1 所示,括号中为该步骤所用软件。3 个平台测序获得的reads 即为原始数据,将各平台各样本的两个文库数据合并后进行后续分析。原始数据质量由fastp[14]统计,质控和adapter 接头去除由Trim Galore[15](0.6.1)执行,所用参数为“--stringency 3 --length 20 -e 0.1”,序列比对由BWA[16](0.7.17)的mem 算法处理,所用参数为“-t 6 -M–R "@RG ID:id LB:id PL:ILLUMINA SM:id"”(其中id 为自定义的样本编号),参考基因组版本为Sscrofa11.1。比对后的sam文件用GATK[17](4.0.12.0)的ReorderSam 排序后由Samtools[18](1.9)转为二进制格式bam 文件,随后用GATK 的SortSam、MarkDuplicates 依次进行排序和重复reads 标记工作,重复reads 只进行标记而不剔除(--REMOVE_DUPLICATES false),随后用GATK的BaseRecalibrator 获取碱基质量校正的校准表文件,“--known-sites”所用dbsnp 库版本为150,另一参数为“--bqsr-baq-gap-open-penalty 30”,最后用GATK的ApplyBQSR 利用上述校准表文件对碱基质量进行校正,获得最终的bam 文件。根据此bam 文件评价比对质量,Samtools 的flagstat 模块用于统计双端reads 比对率,插入片段由Picard[19]的CollectInsertSizeMetrics统计,平均比对深度和位点覆盖超过10X、20X 的比例由Mosdepth[20]输出。以上所列参数外的其余参数均为相应软件的默认参数。
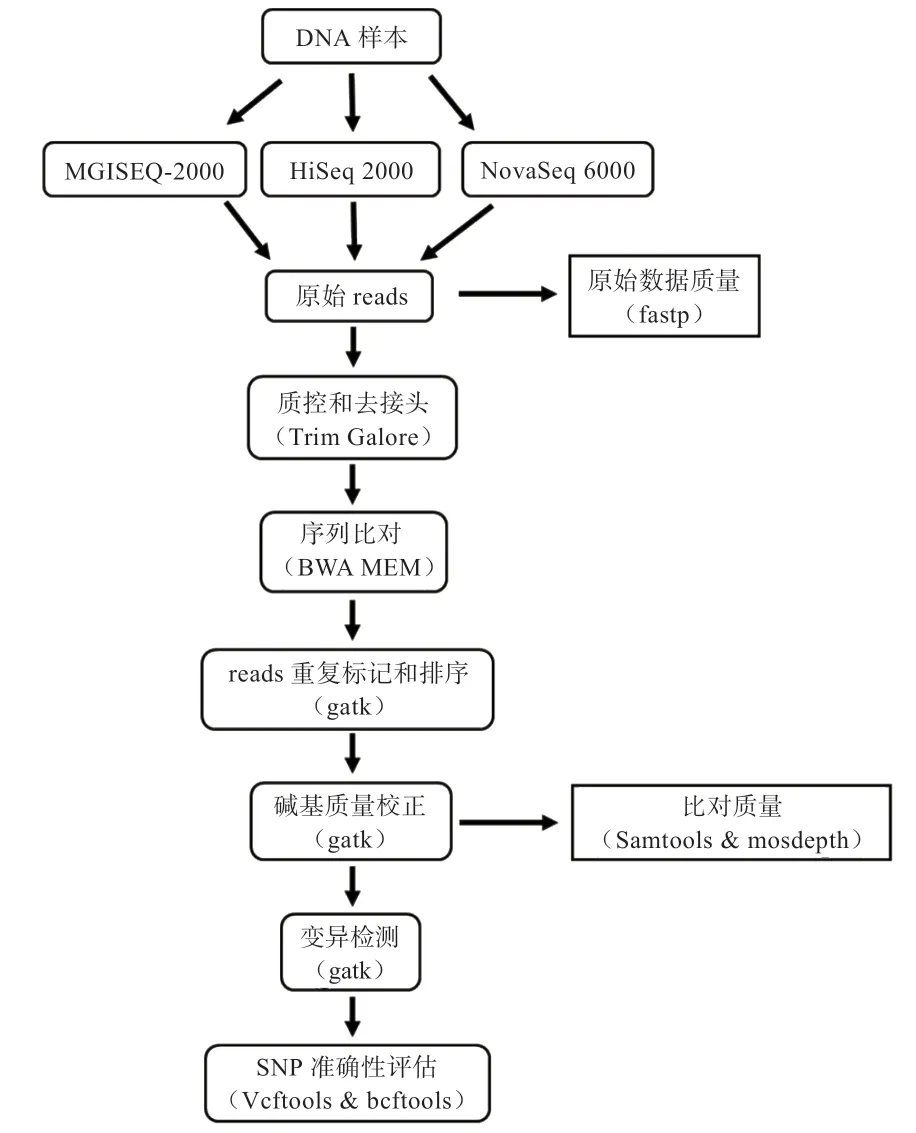
图1 数据分析流程及所用软件
将最终获得的bam 文件用GATK 的Haplotype Caller 进行个体水平的变异检测,使用参数“-ERC GVCF”得到各平台各样本的gvcf 文件,然后分别将各平台各样本的gvcf 文件用GATK 的CombineGVCFs合并,获得3 个平台各包含9 个样本的vcf 文件,再用GATK 的GenotypeGVCFs 分别基于3 个平台各自的vcf 文件进行群体水平的变异检测,得到3 个平台各自的单个vcf 文件,最后用GATK 对检测得到的SNP 和INDEL 执行过滤操作。SNP 的过滤参数为“QD<2.0||M Q<40.0||FS>60.0||SOR>3.0||MQRankSum<-12.5||ReadPos RankSum<-8.0”,INDEL 的过滤参数为“QD<2.0||F S>200.0||SOR>10.0||MQRankSum<-12.5||ReadPosRank Sum<-8.0”。使用VCFtools[21](0.1.16)统计各平台位点数及三者共有位点所占比例,同时用SnpSift[22]对SNP 位点进行注释(本研究中所使用的dbsnp 库版本均为150),然后统计各平台检测的SNP 位点中被dbsnp库收录的位点所占比例。
不同平台结果之间的差异显著性采用SPSS 20.0 的配对样本t检验进行检验,统计检验的显著水平(双侧)设为P<0.05,2 个变量之间的相关系数由Excel 2016中的CORREL 函数计算得出(皮尔逊相关系数)。
1.4 SNP 分型及准确性评价 将50K 芯片(Sscrofa10.2版本)位点坐标在UCSC 数据库转为Sscrofa11.1 版本。Sscrofa11.1 和Sscrofa10.2 中的一些DNA 序列片段是反向互补的关系,找出50K 芯片中位于这些序列上的位点,根据碱基互补原则将其转换,使得测序数据检测SNP 与基因芯片SNP 处于同一条链。随后指定dbsnp库位点的参考碱基(ref)和突变碱基(alt)为基因芯片位点的ref 及alt,用plink[23](1.90)将plink 格式的芯片位点文件转为vcf 格式文件。将9 个样本分型后只存在缺失(./.)和野生纯合(0/0)2 种类型的位点进行剔除。此处的“野生纯合”是指2 个等位基因均与参考基因组碱基一致的位点,这些位点在重测序数据的变异检测过程中未被判定为SNP。各个测序平台vcf 文件中的ref 和alt 与基因芯片vcf 文件中一致,可以通过直接比较位点基因型是否一致来判断测序数据SNP 判型是否正确,即测序平台检测SNP 和基因芯片SNP 二者在某个位点上均为0/0(或0/1、1/0、1/1 三者之一)时,认为该测序平台在该位点上判型正确,判型一致的位点数与位点总数的比值作为SNP 检测准确性的评价指标。
2 结果
2.1 原始数据质量 各平台重测序后的原始数据统计结果见表1。本研究中3 个平台测序后的原始数据量介于61.9~83.9 Gbp 之间,平均测序数据量均在70 Gbp 以上,符合测序深度20X 以上的要求,且3 个平台之间的原始数据量无显著差异。GC 含量介于42%~46% 之间,与参考基因组的42% 相近,表明测序过程中出现序列偏向的可能性较低。NovaSeq 6000 的Q30 以上reads所占比例为91.71%,略高于HiSeq 2000(91.46%)(P>0.05),且二者均高于MGISEQ-2000(86.39%)(P<0.01)。HiSeq 2000 和NovaSeq 6000 的重复reads比例分别为17.17% 和14.57%,高于MGISEQ-2000(0.51%)(P<0.05)。

表1 原始reads 数据量及质量(平均值±标准差)
图2 为3 个平台测序数据中不同位置碱基的质量值分布。HiSeq 2000 和NovaSeq 6000 平台之间的碱基质量值分布的差异较小,而MGISEQ-2000 平台不同位置的碱基质量稍低于其他2 个平台,且波动范围较大,3 个平台reads 不同位置的碱基质量值均在30 以上。虽然3 个平台的原始测序数据质量存在一定差异,但都达到后续分析的要求。
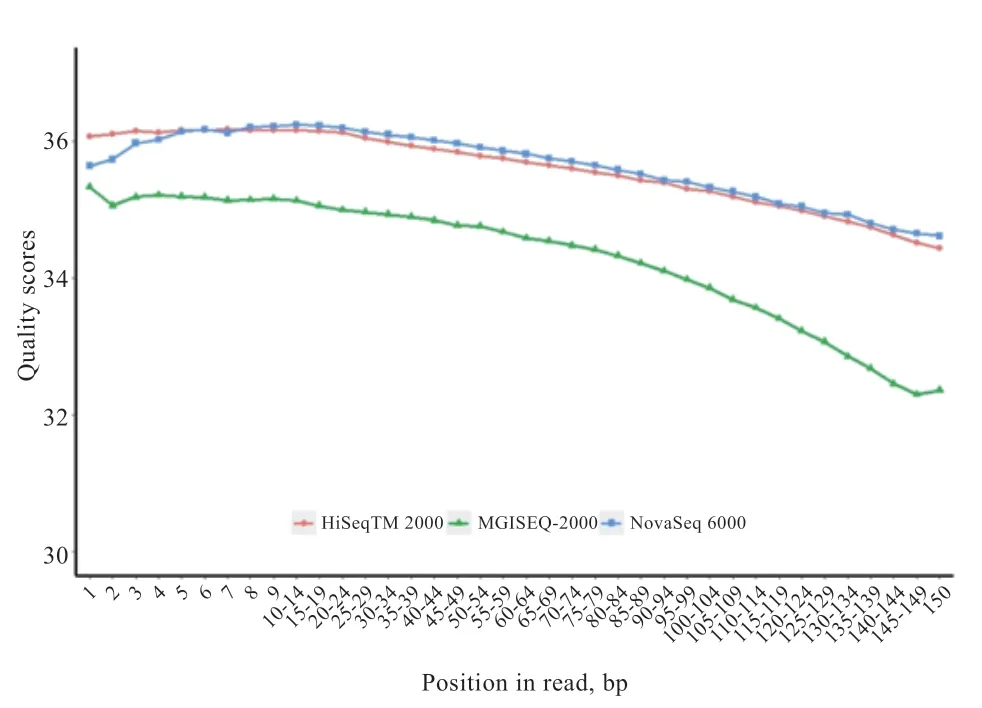
图2 reads 不同位置上的碱基质量分布
2.2 比对质量 MGISEQ-2000 的平均双端比对率为96.20%,高于HiSeq 2000(95.49%)和NovaSeq 6000(95.37%)(P<0.01),后两者间的差异不显著。从图3 可以看出,MGISEQ-2000 和HiSeq 2000 的结果一致性较高(相关系数r=0.94),而NovaSeq 6000 各个样本之间的比对率差异较大,测序稳定性较差。图4 为统计的插入片段长度。3 个平台的平均插入片段长度介于322~382 bp 之间,且较为集中,能在一定程度上反映出建库质量较好。

图3 双端reads 比对到参考基因组的比例

图4 平均插入片段长度
3 个平台的平均比对深度和位点覆盖深度超过20X 的位点所占比例见图5。根据比对结果,3 个平台的平均比对深度均超过20X,达到送测要求。MGISEQ-2000 的平均比对深度为27.35X,高于HiSeq 2000(23.10X)和NovaSeq 6000(24.44X)(P<0.05),而Illumina 2 个平台的平均比对深度无显著差异。MGISEQ-2000、HiSeq 2000 和NovaSeq 6000 覆盖深度在10X 以上的位点比例均在99%以上,分别为99.56%、99.45%和99.59%。3 个平台在覆盖深度超过20X 的位点比例的结果差异较大,MGISEQ-2000 为82.78%,高于其他2 个平台(P<0.05),而NovaSeq 6000(73.11%)高于HiSeq 2000(65.11%)(P<0.05)。

图5 平均比对深度和覆盖>20X 位点比例
2.3 SNP 变异检测 进行群体水平的变异检测后获得3个vcf 文件(每个平台1 个),各平台的变异检测情况见表2。3 个平台所得到的SNP 位点数相似,与Kang等[24]用13 头猪的样本(10 个品种)在HiSeq 2000 平台检测得到的结果2 812 万相当。在SNP 和INDEL 数量上,MGISEQ-2000 多于其他2 个平台。3 个平台的Ti/Tv 均值均为2.40,与Lee 等[25]的研究结果相似。3个平台共有SNP 位点数为27 359 678 个,在各个平台位点总数的占比均达到95% 以上,检出位点一致性较高,另外3 个平台所检测SNP 中dbsnp 库收录位点比例均达到80%以上。
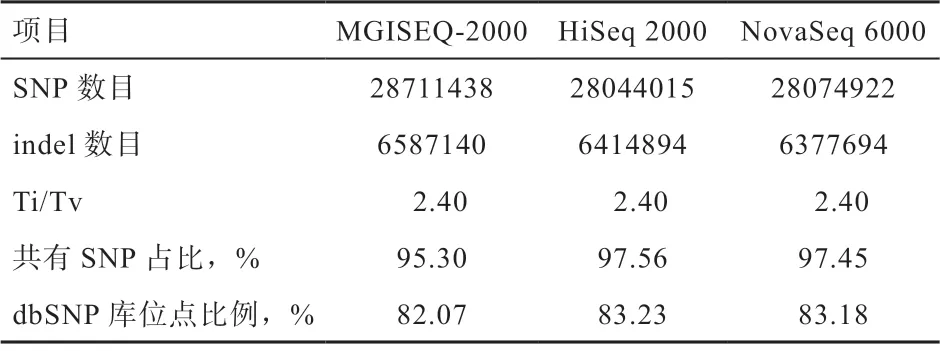
表2 不同平台测序数据变异检测结果统计
2.4 SNP 判型准确性 将参考基因组为Sscrofa10.2 的50K 芯片转为Sscrofa11.1 版本的过程中,有1 677 个位点未在Sscrofa11.1 中匹配到,同时将缺失和野生纯合位点剔除后剩余35 871 个位点。3 个平台各个样本的SNP 判型准确性见图6。MGISEQ-2000、HiSeq 2000和NovaSeq 6000 检出50K 芯片中SNP 位点的比例分别为97.50%、97.43% 和97.40%,MGISEQ-2000 检出的50K 芯片位点数高于其他2 个平台,而Illumina 的2个平台之间结果相近。以基因芯片的判型结果为参考标准,MGISEQ-2000 和HiSeq 2000 的平均准确性均达到97.21%,且二者各样本的准确性高度一致(r=0.94),NovaSeq 6000 的S1~S7 样本的准确性与其他2 个平台相似,但S8 和S9 的准确性较低,分别为77.06% 和76.85%。NovaSeq 6000 的S8 和S9 2 个样本判型与芯片不一致的位点中,2 个样本同时出现错判的位点约占50%。这些位点中,MGISEQ-2000 和HiSeq 2000与芯片基因型一致而NovaSeq 6000 与芯片不一致的位点占90% 以上(S8 为90.20%,S9 为90.94%),MGISEQ-2000、HiSeq 2000 和芯片阵列中判型为纯合位点而NovaSeq 6000 中判型为杂合位点的位点占87%以上(S8 为87.78%,S9 为90.29%)。

图6 3 个平台各个样本的判型准确性
为了分析S8 和S9 样本位点判型错误的原因,本研究选择了S8 样本的一个SNP 位点(chr1:252 645)进行了分析。该位点在基因芯片、MGISEQ-2000 和HiSeq 2000 中基因分型均为T/T,而NovaSeq 6000中基因分型为T/G。在NovaSeq 6000 中共有26 条reads 比对到覆盖该位点的区域,其中21 条在该位点的碱基为A/T(正/ 反链),其余5 条碱基为G/C。在NovaSeq 6000 的S8 个体的最终比对文件中找出该位点碱基为G/C 的reads(共5 条,read1~5),同时选择了该位点碱基为T/A 的reads 作为参考(共2 条,read6~7),用BLAST[26]软件将以上得到的7 条reads比对到参考基因组(Sscrofa11.1)。7 条reads 均比对到了1 号染色体,且完全匹配的碱基在148 个及以上,比对结果可信且与BWA 软件一致,可排除软件比对算法不同带来的差异。图7 展示了该位点(chr1:252 645)前后30 bp 的比对情况,其中横线表示read 在该位点的碱基与第一行中的参考序列对应位置的碱基一致,同时显示了所有reads 在1 号染色体252 645 位置上的碱基。S8 在该位点出现G 等位基因即由read 1~5 引起,可判断该判型错误出现在原始reads 上,测序错误造成了SNP 的分型错误。

图7 chr1:252 645 的局部比对情况
3 讨 论
3.1 原始数据质量 Q30 以上reads 比例不仅受样本类型、文库质量、插入片段长度等因素影响,还与测序试剂和光信号采集过程等因素有关。虽然本研究中MGISEQ-2000 的Q30 以上reads 比例低于HiSeq 2000和NovaSeq 6000,但其已远远超过了该平台宣传手册上Q30>75% 的性能参数[7]。Korostin 等[10]的研究中华大智造的MGISEQ-2000 的Q30 以上reads 比例同样低 于Illumina 的HiSeq 2500 平 台。MGISEQ-2000 测序重复率显著低于Illumina 平台的原因可能是其采用了CoolMPS 测序试剂套装和序列片段线性扩增的建库方式。本研究只对重复reads 进行了标记而未将其删除,相关研究发现测序分析过程中是否去除重复的reads 对后续分析影响不大[27]。虽然3 个测序平台在原始测序数据上表现出一定的差异,但三者均达到了测序要求,可以进行下游的数据分析。
3.2 序列比对质量 从表1 中可以看到,NovaSeq 6000在原始数据量、Q20 及Q30 以上reads 比例上均高于其他2 个平台,但其在双端比对率上却低于后两者。另外,在图3 中可以看到,NovaSeq 6000 各个样本之间的双端比对率差异较大,而其他2 个平台差异较小且不同样本之间变化一致,表明NovaSeq 6000 的测序稳定性不 如MGISEQ-2000 和HiSeq 2000。NovaSeq 6000 在平均比对深度和覆盖深度大于20X 的位点比例上高于HiSeq 2000,但低于MGISEQ-2000(P<0.05),但应注意3 个平台测序数据量的差异给比对质量评价带来的影响。在本研究中,MGISEQ-2000 虽然在原始数据量上少于其他2 个平台,但其比对质量却好于Illumina 的HiSeq 2000 和NovaSeq 6000 平台。以上结果说明原始测序数据的数据量和Q20、Q30 等不能直接反映比对质量,所以在选择测序服务时可以对序列比对环节的质量做出要求,进一步保证测序数据质量。
3.3 变异检测结果 在变异检测上,本研究主要分析了SNP 和INDEL 2 种变异类型。HiSeq 2000 和NovaSeq 6000 2 个平台不仅在变异检测数目上非常接近,其共有位点和dbsnp 库收录位点比例也均高于MGISEQ-2000,表明Illumina 不同时期发布的测序平台仍能保持较高的一致性。而MGISEQ-2000 变异检测数目高于其他2 个平台,这可能与其平均比对深度和覆盖深度在20X 以上的位点所占比例高于其他2 个平台有关。总体上看,MGISEQ-2000、HiSeq 2000 和NovaSeq 6000 检测出的SNP 和INDEL 变异数目较为接近。
3.4 SNP 判型准确性 在与基因芯片位点比较的过程中,3 个平台都存在未检出SNP 芯片中所有位点的情况,原因可能是测序深度不够,一些杂合位点未被检出,相关研究发现,重测序数据检测出所有杂合位点要求测序深度在33X 以上[12]。实验所用的猪为中国地方品种(马身猪和大河猪),而比对使用的参考基因组(Sscrofa11.1)为杜洛克品种,可能一些地方猪种的特异位点不能被检测到,但通过分析发现实验中马身猪和大河猪两者在双端比对率、平均比对深度和覆盖>20X 的位点比例上无显著差异。在基因分型结果上,MGISEQ-2000与HiSeq 2000 位点基因型都与芯片位点高度吻合,而NovaSeq 6000 除S8 和S9 外的样本也与芯片结果一致。本研究中剔除了SNP 芯片中只由野生型纯合和缺失2种类型组合的位点,这些位点因在群体内基因型一致,在变异检测中未被判定为SNP,所以其实际上与芯片位点基因型是一致的,这使得本研究中的重测序数据检测的SNP 与基因芯片的共有位点数和判型准确性偏低。部分重测序检测SNP 位点的基因型与SNP 芯片不一致,表现形式为测序检出的SNP 为缺失或多等位基因,可能的原因是该SNP 位点周围存在INDEL 等变异,导致该位点上的碱基移位或缺失。NovaSeq 6000 平台S8 和S9 样本的原始数据量均在72 Gbp 以上,达到Q30 以上的reads 比例也均在91%以上,且两者的双端比对率分别为71%和64%,与S1~S7 样本类似,所以在原始数据质量和比对质量上不能发现这2 个样本存在问题。从NovaSeq 6000 的S8 样本的局部比对结果中可以看出,判型错误的原因在于测序过程中出现了碱基错判,其原因可能是DNA 延伸时连接了错误的碱基或者是光信号采集中出现了误读,可以考虑进一步采用PCR 和Sanger 法测序验证这些位点的准确性。
4 结 论
MGISEQ-2000 在重复reads 比例和比对质量方面均优于HiSeq-2000 和NovaSeq-6000,在SNP 变异检测的准确性上与HiSeq-2000 相当且高于NovaSeq-6000。NovaSeq 6000 在原始数据和序列比对上优于HiSeq 2000,而在SNP 检测准确性上低于HiSeq 2000,且存在测序上的系统性误差。综合而言,HiSeq-2000 的测序质量与近几年推出的二代测序相比未表现出明显差距,而MGISEQ-2000 平台重测序表现性能稳定、质量可靠,在实际应用上有明显的优势和应用价值。
