炎症因子在骨质疏松发病机理中的研究进展
2021-11-11李崇罗晓婷纪舒妤谢天裕李剑林成森李波香黄能干阳富春刘云
李崇 罗晓婷 纪舒妤 谢天裕 李剑 林成森 李波香 黄能干 阳富春 刘云*
1.广西医科大学第一附属医院骨科,广西 南宁 530021 2.广西医科大学第一附属医院药学部,广西 南宁 530021 3.广西医科大学第一附属医院全科医学病区,广西 南宁 530021
骨质疏松症(osteoporosis, OP)是一种以骨密度(bone mineral density, BMD) 降低和骨量丢失、骨微结构破坏的全身代谢性骨病,以骨脆性增加、易发生骨折为主要特征[1-2]。随着人口老龄化增加,全球约2亿人口受此困扰,老年人的OP发病率、损伤率和致残率逐年升高,严重影响患者生活质量,给家庭及社会造成巨大负担[3]。
生理性的骨骼稳态由成骨细胞(osteoblast,OB)的骨形成和破骨细胞(osteoclast, OC)的骨吸收来维持相对稳定的平衡,而OB与OC作用失衡是OP发病关键。大量研究表明IL-1、TNF-α等炎症因子对OB与OC的活性与功能有重要影响,在骨质疏松的发病中扮演重要角色[4]。本文根据炎症因子对OP发挥作用机制不同分为促进、抑制、双作用,并分别进行归纳总结。
1 炎症因子概述
炎症因子是由免疫细胞及组织细胞(如淋巴、单核/巨细胞等)分泌、具有调控作用的一类小分子可溶性分泌蛋白(8~30 kD),通过自分泌、旁分泌或内分泌的方式实现细胞间信息交流来调节免疫和炎症反应[5]。炎症因子按来源主要分为两种:白介素(interleukin,IL)、肿瘤坏死因子(tumor necrosis factor, TNF);按功能可分为三类:①促进OP发生的炎症因子,如IL-1、IL-17和TNF-α;②抑制OP发生的炎症因子,如IL-10;③具有促进和抑制OP双重作用的炎症因子,如IL-6、IL-8。炎症因子的释放不仅会影响免疫细胞的产生活化,还可互相作用,在较低水平下仍具有生物学活性,从而能有序进行分工合作并协调功能,也易触发信号级联反应[6]。
1.1 促进OP发生的炎症因子
大部分炎症因子如IL-1、TNF-α等,主要是激活或抑制RANK/RANKL/OPG、Wnt等信号通路,进而引发促进OC分化或抑制骨髓间充质干细胞(MSCs)成骨分化等生物效应,使得骨吸收增加及骨形成减少导致骨量丢失。
1.1.1IL-1 :IL-1主来源中性粒细胞、单核-巨噬细胞等,分为:IL-1α和IL-1β。绝经后OP患者IL-1水平升高且与BMD呈负相关[7]。IL-1α可上调巨噬细胞集落刺激因子(M-CSF)和前列腺素E2(PGE 2)的表达介导骨质疏松的发生[8],此外,IL-1β、TNF-α、IL-6联合作用可增强OC活性[9]。
在作用机制方面,IL-1通过激活NF-κB受体活化因子(RANK)/NF-κB受体活化因子配体(RANKL)/骨保护素(OPG)信号通路介导OC生成并抑制其凋亡,导致骨吸收增加,破坏骨吸收与骨形成之间的平衡,促进骨质疏松的发生发展[10-11]。IL-1α及IL-1β均可诱导OB表面的RANKL表达增加,OPG表达减少[12-13],RANK与RANKL结合可促进OC的激活与分化并抑制成熟OC的凋亡,而OPG作为RANKL的竞争性抑制剂,其表达下降进一步导致骨吸收增加[14]。
此外,IL-1能通过调节Wnt通路促进OP进展,其上调Dickkopf-1(DKK-1)、硬化蛋白(Wnt蛋白拮抗剂)表达而抑制OB活性,导致骨形成减少[15]。
目前,IL-1在OP发病机制中的作用虽已有初步研究成果,但其深入的机制尚未完全清楚,如IL-1是通过干预何种信号通路来调节RANKL和其他信号分子的表达,研究其作用机制可为OP的诊断及相关干预药物的研发提供新思路。
1.1.2IL-17:IL-17通常是指IL-17家族中的IL-17A,主要由T辅助细胞17(Th17)产生,是促炎细胞因子[16]。最近研究表明,绝经后OP患者血清IL-17水平显著增加,且IL-17A与BMD呈负相关[17],说明IL-17可导致骨质流失,从而参与OP的发生。
IL-17主要通过影响OC及OB分化促进OP发生发展。一方面IL-17可上调神经型N-粘钙蛋白(N-cadherin)表达、抑制Wnt信号转导从而抑制OB分化和骨形成[18]。IL-17还可激活IKK-NF-κB通路抑制MSCs的OB分化。此外,IL-17能上调hBM-MSCs中瘦素的表达,从而抑制JAK / STAT通路介导的脂肪形成进而抑制其成骨分化[19-20],说明IL-17抑制MSCs向OB分化介导OP发生。
另一方面,IL-17直接刺激OC分化,其作用取决于OC前体的来源和亚型[21]。低水平的IL-17A通过RANKL-JNK1途径促进OC前体的自噬活性[16],从而促进OC分化和增强其活性。此外,IL-17作用于OB使PEG2分泌增加、OC分化因子表达增加而促进OC的分化成熟[22],导致骨吸收增强及骨量流失。IL-17还能上调IL-1β、IL-6、IL-8、TNF-α等因子的表达,从而协同放大炎症反应[23],调节OC的NF-κB和RANKL的分泌。研究示抗IL-17治疗可抑制促炎因子、促进抗炎因子的产生,其效果比较抗RANKL和抗TNF-α的效果好[24]。
由此可见,IL-17对OP的影响非常显著,目前抗IL-17及抗IL-17抗体治疗已有不少研究,其有望成为治疗OP的高效药物。
1.1.3TNF :TNF是对肿瘤细胞具有细胞毒作用并能使肿瘤组织缺血坏死的细胞因子,由Carswell等[25]于1975年首次提出,根据来源及结构分为TNF-α和TNF-β。TNF-α是由巨噬细胞、T与B细胞等分泌形成的同三聚体,与TNF-RI(TNFRSF1A或p55)和TNF-RII(TNFRSF1B或p75)受体结合而发挥作用[26]。例如,TNF-α能直接与破骨前细胞上的TNFRI结合促进OC的骨吸收[27]。目前已有许多报道TNF-α可通过促进OC的骨吸收且抑制OB的骨形成作用而参与OP发生。
TNF-α能诱导TNF超家族的成员之一的RANKL分泌增加,而RANK和RANKL结合后募集TNF受体相关因子(TRAFs,多为TRAF6),后者激活NF-kB、MAPK和AKT等信号通路,增强OC的活性及骨吸收作用[27-28]。Li 等[29]研究发现TNF-α可通过激活PI3K / Akt信号通路而促进RANKL诱导OC形成的协同作用。TNF-α还可激活JNK信号通路上调Semaphorin3D的表达,介导破骨前细胞增殖及OC的分化[30]。此外,有研究[31-32]发现TNF-α可诱导OC的let-7a的高表达,后者通过转录后抑制Fas/FasL信号通路的分子表达,从而抑制OC凋亡以促进OP的发生发展。
此外,TNF-α通过抑制MSCs的增殖和成骨分化来抑制骨形成,其不仅能抑制Wnt信号通路抑制MSCs的成骨分化,还能上调SOX5、泛素连接酶(SMURF1)的水平及下调Semaphorin3B介导的信号通路抑制MSCs的成骨分化[33-35]。Liao等[36]发现TNF-α激活 NF-κB信号通路后促进miR-705表达,降低MCSs中FoxO1蛋白的水平,FoxO1水平降低导致Ros水平升高,Ros的升高进一步激活TNF-α及NF-kB信号通路而形成正反馈通路,故当FoxO1水平降低后导致MCSs的抗氧化防御能力下降,使得MSCs的OB分化受抑制及OB数量减少而导致OP发生。
TNF-α的GG基因型也可造成绝经后OP患者骨密度下降[37],TNF-α诱导的miR-204表达对MSCs成骨分化的抑制作用已有研究报道[38],相反,miR-216a则具有促进作用[39]。TNF-α可通过诱导miR-23b表达而显著降低了Runx2的水平,从而间接抑制了MSCs的成骨分化[40]。
综上所述,TNF-α主要通过介导多种信号通路来发挥促OP作用,探寻其发病机制可为寻找特定的信号通路及治疗靶点而阻断TNF-α促进骨吸收和抑制骨形成的作用提供研究支持,然而有研究报道高盐饮食可使小鼠TNF-α水平升高而致骨质流失[41],因此是否可通过干预生活方式预防OP的发生也值得今后去探索。
1.2 抑制OP发生的炎症因子
在OP发生过程中,亦有极少数炎症因子发挥负调控作用,如IL-10,其通过调节NFATc1、OPG、RANKL等的表达而激活相应的信号通路,使OC产生减少、OB标志物表达减少等,延缓OP进程。
IL-10由单核巨噬细胞和Th细胞等合成分泌的、主要参与炎性与免疫反应的多效细胞因子,已被公认为炎症和免疫的抑制因子,属于少有的炎症抑制性细胞因子[42],OP患者IL-10表达水平较低,其可由OC分泌至周围的细胞,并通过负反馈机制来抑制OC的分化[43]。
Claudino 等[44]发现在缺乏IL-10的情况下,牙槽骨的骨量丢失主要与OB和骨细胞标志物的表达减少、OB的增殖分化受抑制有关,而与微生物、炎症或骨吸收途径无关,IL-10敲除后骨量丢失增加、小梁结构改变、骨密度降低[45]。
在作用机制方面,IL-10直接作用于单核细胞前体,抑制NFATc1的表达和核易位,激活JAK2/STAT3通路,诱导PD-L1的表达,使Ca2+移动中断从而抑制OC的分化[46]。此外,IL-10还可上调OPG的表达、下调RANKL和M-CSF的表达,再通过TRAF6进一步抑制(TRAF6是泛素连接酶,可作为开关激活信号通路)NFATc1的激活,进而抑制OC的激活[47]。IL-10也可通过抑制TGF-b信号而减少Runx2表达量来抑制OC的分化[48]。最后在方丹青等[49]发现当IL-10升高时,NF-kB蛋白的表达量会相应减少,而NF-kB信号通路能促进OC的分化,由此亦证实了IL-10对OC分化的抑制作用。总之,IL-10调节OC的机制可能与调节NFATc1、OPG、RANKL、TGF-b和NF-kB等基因表达量有关。
虽然IL-10抑制OC分化的具体信号通路、关键因子及相关作用机制仍不清楚,但在抑制OP发生发展中其重要作用,有望成为未来治疗OP的新药物。
1.3 对OP发生具有双重作用的炎症因子
有些炎症因子具有促进和抑制OP发生的双重作用,如IL-6、IL-8 等。两者促进OP发生的主要机制同上述,但在抑制OP方面却有所不同,它们在OP发生过程中发挥着总体促进的效应。
1.3.1IL-6 :IL-6来源基质细胞、OB等[4],属于促炎细胞因子。OP的发生伴随血清IL-6水平升高,抗IL-6受体抗体能抑制骨吸收并增强骨形成[50]。另外,IL-6的基因高表达增加了OP的风险,且IL-6基因还通过基因-基因的作用在OP疾病进展中发挥作用[51]。IL-6必须先与膜结合受体(mIL-6R)或可溶性受体(sIL-6R)结合形成复合物,再与IL-6 的信号传递链(glycoprotein 130,gp130)结合形成二聚体才能激活其诱导的信号通路[52]。然而,mIL-6R的作用范围小,故IL-6的生物学效能主要通过sIL-6R实现,且IL-6仅通过与sIL-6R结合后的信号转导起促进OC生成和骨形成的作用[53]。IL-6与sIL-6R结合后可与OB膜融合,激活JAK/STAT3,导致RANKL、IL-1、甲状旁腺素相关蛋白(PTHrP)和PGE2水平升高[54],PTHrP的升高会引起PTH的升高,而PTH、1,25-二羟维生素D3[1,25(OH)2D3]可促进IL-6、RANKL的产生[55],且PGE2也可促进IL-6的产生,并抑制OB表达OPG。故形成了一个正反馈通路使得RANKL/OPG的比值增大,从而促进OC的增殖与分化,加速骨吸收。
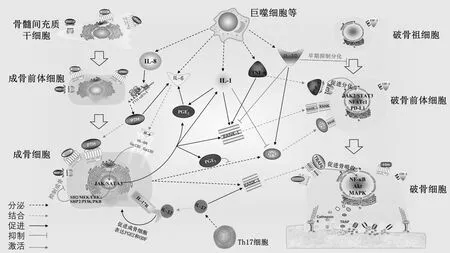
图1 炎症因子IL-1、IL-6、IL-8、IL-10、IL-17、TNF-α六种炎症因子在骨质疏松症发病中的机制图Fig.1 The mechanism of six inflammatory factors IL-1, IL-6, IL-8, IL-10, IL-17, and TNF-α in the pathogenesis of osteoporosis
此外,IL-6与sIL-6R结合还可激活SHP2/MEK/ERK 与SHP2/PI3K/PKB通路,从而抑制OB的骨形成并降低OB矿化作用[56],加之IL-6和sIL-6R均可下调与OB相关基因的表达量,如碱性磷酸酶等[57],导致骨形成减少。然而,在王信等[58]研究中,IL-6能直接抑制经RANKL诱导的单核巨噬细胞向破骨细胞分化,从而降低OC的骨吸收效应,表明IL-6能通过抑制NF-κB信号通路抑制OC的分化成熟和骨吸收效应。
总的来说,IL-6在骨质疏松的进程中表现出的综合效应为促进骨吸收。在治疗方面,若能通过对IL-6不同的基因位点进行基因型检测以及阻断其介导的多种信号通路的特定靶点,有可能达到预测OP发生的风险和早期预防诊治OP的目的。
1.3.2IL-8 :IL-8由巨噬细胞、上皮细胞等产生,可激活p38 MAPK / ERK-NF-κB通路介导炎症反应[59]。IL-8表达水平随着年龄的增加而增加,是老年人骨量丢失的重要原因,且在OP骨折患者的OB培养中也可以检测到恒定的 IL-8 表达。其机制包括IL-8促进PTH的合成,从而抑制OB的骨形成作用,促进单核细胞分化为OC,增加骨质吸收[60]。此外,IL-8可以通过上调OC中基质金属蛋白酶(MMP-9)的表达,后者的表达量可随着IL-8的水平、作用时间的增加而增加,进而加快OC的分化速度及程度,最终加剧骨质的流失导致OP[61]。
在抑制OP方面,IL-8可通过抑制OC的骨吸收,也促进软骨转录因子和标志物(如 Sox9和II型胶原)的表达、激活HIF-1a通路而促进骨生成来抑制OP进程[62-63]。
IL-8对骨质疏松的影响具有双重效应,大多数学者认为其水平的升高,容易导致OP,主要机制是IL-8可升高PTH水平,并可上调OC中MMP-9表达。但也有学者认为IL-8可以刺激OC的运动而抑制骨吸收作用,并通过HIF-1α通路促进OB骨形成。
1.4 其他炎症因子
Amcheslavsky等[64]研究表明,IL-12既可通过降低NFATc1水平直接抑制OC的形成,又可抑制RANKL诱导的OC分化;IL-23通过上调RANK表达促进OC的形成[65],但对OB形成和RANKL的表达无影响[66]。有研究[66]表明,IL-23和IL-27可部分抑制RANKL诱导的BMMs分化形成OC,而不影响其增殖。此外,IL-27还可以促进OB的STAT3激活而诱导RANKL和OC生成。IL-35可激活JAK1/STAT1信号通路而抑制TNF-α和MAPK的表达,进而促进OC凋亡,起到延缓OP发生的作用。
2 总结与展望
本文初步介绍IL-1、IL-6、IL-8、IL-10 、IL-17、TNF-α等常见的重要炎症因子,通过激活或抑制相关的信号通路、调节其他细胞因子的表达量、基因多态性等不同的方式参与OP的发生、发展过程,但具体机制尚未明确。目前大多数研究都是针对某一炎症因子对OP发病机制中的某一环节进行的,且主要停留在动物模型或体外实验的研究中,而各种炎症因子共同导致OP发生的综合性研究仍较少,尚不能更深层次从整体上揭示炎症因子在OP发病机制中的作用,对临床工作的指导有限,诸多问题亟待解决。本文通过梳理常见的重要炎症因子在OP发病机制中的作用,进而从病理机制的角度重新认识OP与炎症因子的关系,这有助于能否将炎症因子作为临床生物指标纳入管理中,对于OP的早期预防、诊断、治疗以及预后评估等具有重大意义,尤其为寻找有效抗OP方案提供理论基础。
