Bioinformatics: A beacon of hope in identifying molecular target
2021-11-08KngChunWngZhengGungWngYiBoDiHngFeiWuDeHuiYi
Kng-Chun Wng ,, Zheng-Gung Wng , Yi-Bo Di , Hng-Fei Wu ,, De-Hui Yi ,∗
a Department of Organ Transplantation and Hepatobiliary, The First Affiliated Hospital of China Medical University, Shenyang 110 0 01, China
b The Key Laboratory of Organ Transplantation of Liaonin g Province, The First Affiliated Hospital of China Medical University, Shenyang 110 0 01, China
c Department of Orthopedics, The First Affiliated Hospital of China Medical University, Shenyang 110 0 01, China
d Northeast Yucai School, Shenyang 110 0 01, China
Hepatobiliary malignancies are highly heterogeneous with poor prognosis and drug response [1] . It is difficult to diagnose at an early stage. Hence, precise molecular targets are urgently needed to improve the diagnose efficacy and individual treatment re-sponse. However, reliable biomarkers that can predict the individ-ual treatment response are still lacking. With the broad applica-tion of high-throughput sequencing technology and bioinformat-ics analysis in scientific research, increasing numbers of diagnos-ing molecular targets will be found and tested. Such bioinformat-ics will provide a more rational way to diagnose, prevent, and treat hepatobiliary cancers [2] .
Bioinformatics, as a cutting-edge technology characterized by innovative technology and high-throughput omics, has become an important method for researchers to screen and identify effective molecular targets in recent years. Bioinformatics mainly refers to the using informatics, statistics, and computer science to study biological issues. As a multidisciplinary research method, it has also been developed by leaps and bounds with the advancement of computer technology in recent years [3] . The novel molecular biomarkers that can predict responsiveness to immune-checkpoint blockade are being extensively investigated to further improve pre-cision immunotherapy [4] . In the studies of hepatobiliary malig-nancies, many researchers take bioinformatics analysis as the start-ing line for research, acquiring and utilizing the complex biological information to explore new target genes or pathways in a certain disease, and then further experimentation is conducted to investi-gate whether those variants can serve as novel biomarkers to ad-dress liver and biliary carcinoma system disparities, which save a lot of money and resources.
In recent years, bioinformatics-related papers in hepatobiliary diseases, especially in the tumor field, continue to increase. We have collected 1813 bioinformatics articles related to hepatobiliary diseases in the past 20 years on the Web of Science, and we ana-lyzed the data using the R package “bibliometrics”. We found that the number of publications has shown a significant upward trend in the past two decades. Of note, a total of 10 0 0 articles have been published in the last four years, which exceeds the total amount of articles published in 2001-2016 ( Fig. 1 ). Besides that, China, USA, and Germany are the top 3 most productive countries ( Fig. 2 ). Compared with Europe and North America, hepatobiliary cancer has a higher incidence in East Asia, and China has the largest num-ber of hepatitis B and liver cancer patients in the world, which is one of the reasons why researchers are so interested in this field.
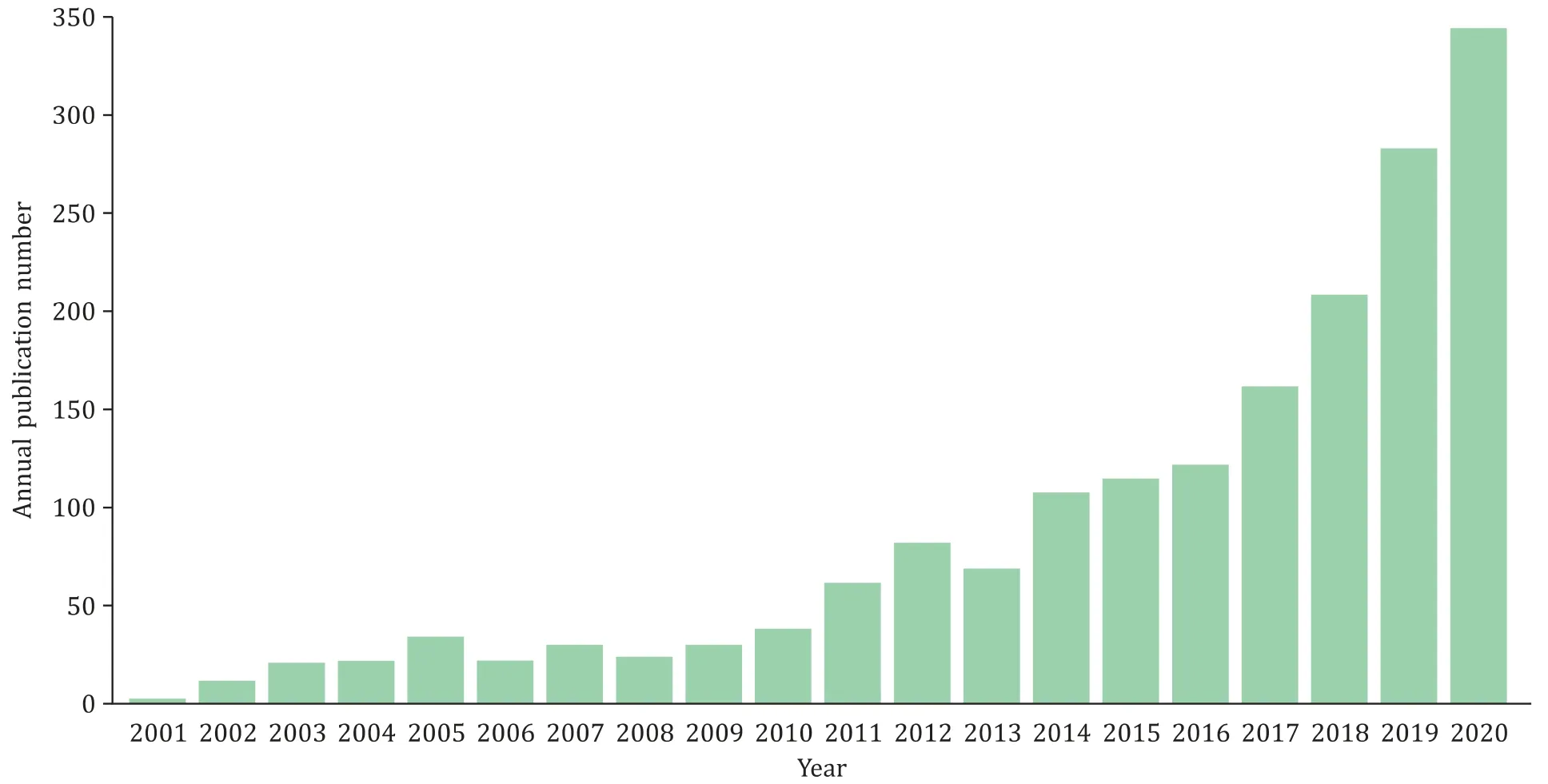
Fig. 1. Annual publication number of bioinformatics-related papers in hepatobiliary field from 2001 to 2020.
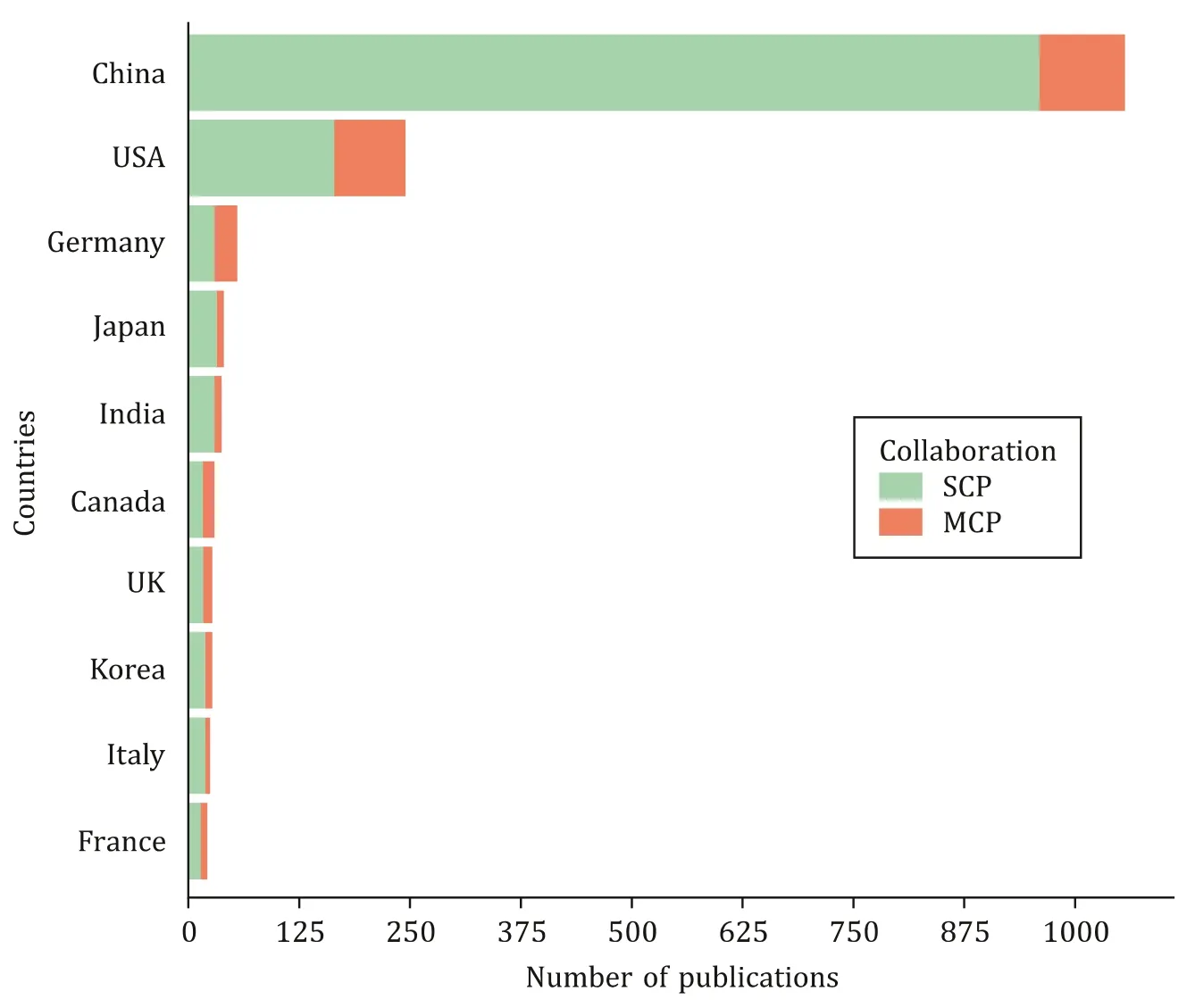
Fig. 2. Most productive countries from 2001 to 2020. SCP: Single country publications; MCP: multiple country publications.
The data sources of bioinformatics analysis mainly include the following aspects, researchers collected clinical tissue samples and obtained expression of genes through high-throughput sequencing technology. On the other hand, the open-access public databases, such as the gene expression omnibus (GEO) database ( https://www.ncbi.nlm.nih.gov/geo/ ) and The Cancer Genome Atlas (TCGA) database ( https://portal.gdc.cancer.gov/ ) published by the National Center for Biotechnology Information (NCBI), is another choice. Besides, more and more public databases, online analysis web-sites, and R programming language packages are constantly emerg-ing, leading to updates and iterations of analysis methods. Based on this, researchers can screen out differentially expressed genes (DEGs) in tumor tissues quickly and easily, and the clinical infor-mation and survival data of various cancers in these databases can be accessed any time, saving several years of follow-up.
In addition to screening some specific genes, the interaction among genes has also caught people’s eyes in recent years. Many researchers construct the protein-protein interaction (PPI) net-works to explore the interactions of DEGs and identify hub genes, and conduct the pathway and functional enrichment analysis to explain the certain processes involved in the disease. Interaction networks and enrichment analysis provide explanations for the acquired data. Many interaction databases and visualization soft-ware, such as STRING database ( https://string-db.org/ ) and Cy-toscape ( https://cytoscape.org/ ), can perfectly meet people’s re-quirements [5] . Zhang et al. [6] downloaded the original data from the GEO database and screened 1230 DEGs in liver can-cer. In the PPI network, the key gene of progression and mi-gration, guanine monophosphate synthase (GMPS), was identi-fied. Kong et al. [7] constructed the competitive endogenous RNA (ceRNA) network of gallbladder cancer on Cytoscape and screened out two important pathways in the pathogenesis of gallblad-der cancer: XLOC_011309-miR-548c-3p-SPOCK1 and XLOC_012588-miR-765-CEACAM6.
As a fast and efficient analysis method based on computers, bioinformatics will continue to exert a unique influence on sci-entific research, especially life sciences. In the future, bioinformat-ics, together with various modern computer tools, will bring about tremendous changes and far-reaching effects on the research of various diseases including liver and gallbladder diseases.
Acknowledgments
None.
CRediT authorship contribution statement
Kang-Chun Wang:Data curation, Investigation, Visualization, Writing –original draft.Zheng-Guang Wang:resources, Software, Visualization.Yi-Bo Dai:Investigation, Writing –original draft.Hang-Fei Wu:Data curation, Formal analysis.De-Hui Yi:Concep-tualization, Project administration, Supervision, Writing –review & editing.
Funding
None.
Ethical approval
Not needed.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the sub-ject of this article.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Comparison and development of advanced machine learning tools to predict nonalcoholic fatty liver disease: An extended study
- Hepatobiliary&Pancreatic Diseases International
- Influence of weight management on the prognosis of steatohepatitis in chronic hepatitis B patients during antiviral treatment
- Key factors and potential drug combinations of nonalcoholic steatohepatitis: Bioinformatic analysis and experimental validation-based study
- LC-MS-based lipidomic analysis in distinguishing patients with nonalcoholic steatohepatitis from nonalcoholic fatty liver
- Cryptococcosis in patients with liver cirrhosis: Death risk factors and predictive value of prognostic models