核磁共振氢谱定量检测3 种食用菌麦角固醇
2021-11-05傅利军张秀敏马永征徐明芳
傅利军,刘 莉,张秀敏,马永征,孙 勇,,岳 甜,徐明芳,
(1.北京食品科学研究院,北京 100068;2.暨南大学生命科学技术学院,广东 广州 510632)
食用菌是一类口感鲜美、营养丰富且具有食药用价值的大型子实体真菌,它不仅具有高蛋白、低脂肪、低胆固醇和富含8 种人体必需氨基酸等特点[1],还含有多种生理活性物质,包括多糖、酚类、倍半萜、超氧化物歧化酶和甾醇类[2]等,帮助机体发挥抗炎、抗癌、抗氧化、抑菌、肝保护、降血糖和抗糖尿病等作用[3-4],适合于各类人群包括免疫力低下、老年人和糖尿病患者等食用,是公认的绿色健康食品。食用菌富含甾醇类,其中包括麦角固醇。麦角固醇是食用菌细胞膜的重要组成部分,在维持食用菌的细胞膜结构完整和功能正常运行方面起着重要作用,也可作为工业用生产医药类产品[5]。麦角固醇也是麦角钙化甾醇(VD2)的光转化前体。食用菌通过紫外光照处理异构化转化为VD2,得到的富VD2食用菌经由膳食途径摄入,可预防和治疗因VD2缺乏或不足产生的儿童软骨病、佝偻病和老人骨质疏松症等,是人体获取VD2的良好途径。
麦角固醇和VD2的定量检测方法目前已得到广泛研究,多见于高效液相色谱(high performance liquid chromatography,HPLC)法[6-7]、气相色谱法[8]以及各种联用技术[9]等精确定量方法。定量核磁共振波谱(quantitative nuclear magnetic resonance spectroscopy,qNMR)是一种近年来新兴的定量检测方法,它是1935年核磁共振现象被发现以来并用于结构定性方面的进一步发展。其定量基本原理是根据Bloch方程,即核磁谱图中粒子吸收峰峰面积只与待测组分所包含的粒子数呈正比,通过积分谱图目标特征信号峰,即能获得待测组分的含量。目前,随着磁场强度的提高、超低温碳头的出现,其检测的灵敏度、精确度逐渐得到提高,qNMR在食品[10-13]、药品[14-15]、生物[16]、代谢组学[17-19]和化合物纯度检测[20-22]等方多面得到了广泛应用。如刘晓婷等[23]建立固相萃取-qNMR测定板蓝根饮片中的表告依春含量的方法,得出方法的精密度小于1%,可用于低含量复杂样品的定量分析;Tanja等[24]利用核磁共振定量研究当归药材有效成分,利用反相固相萃取将当归活性浓缩成单个组分,再通过碱性磷酸酶、(抗)雌激素活性和细胞毒性实验,建立了定量1H-NMR(qHNMR)方法,对当归内酯等活性组分进行了鉴定和定量,并建立了真实当归制剂的质量标准。Canela-Garayoa等[25]采用单一核磁共振研究生物催化合成乙基脂肪酸酯,检测结果与气相色谱法结果一致。qNMR是一种无损检测技术,对测样品纯度要求不高,样品预处理步骤简单,可用于复杂混合物的定量分析,专属性强,一定程度上弥补了常规色谱检测方法的局限性。随着美国、日本和欧洲药典相继将qNMR收载以及我国药典的收录,其在全世界范围内已被作为一种标准测定方法得以应用。
目前核磁定量方法用于食用菌样品检测主要集中在食用菌三萜类[26]、苯类[27]化合物定量的检测,对于食用菌中麦角固醇的定量检测鲜见报道。本研究将选择C-18H质子单峰作为麦角固醇的目标定量特征峰,建立食用菌麦角固醇的定量检测方法,探讨定量检测麦角固醇的可行性,旨在弥补qNMR在检测食用菌麦角固醇方面的空白,为食用菌的新产品研发与质量标准完善提供理论依据与技术方法。
1 材料与方法
1.1 材料与试剂
每包100 g共3 包食用菌(鸡油菌、茶树菇、虫草)干品购自云南文山农贸市场,阴凉干燥玻璃罐储存备用。
麦角固醇标准品(CAS57-87-4)、VD2标准品(CAS50-14-6) 美国Sigma公司;氘代氯仿(99.8%,含TMS) 上海麦克林生化科技有限公司;吡嗪(99%) 上海源叶生物科技有限公司;无水乙醇(≥99.7%)、石油醚(60~90 ℃) 天津市致远化学试剂有限公司;氢氧化钠、甲醇(均为优级纯) 西陇科学股份有限公司。
1.2 仪器与设备
Avance III 600 MHz型超导核磁共振波谱仪 德国 Bruker公司;5 mm核磁管带压力帽(WG-1000-7) 美国Wilmad公司;1100型液相色谱仪 美国Agilent 科技有限公司;CP224C电子天平 奥豪斯仪器有限公司;ER-182A型电子分析天平 日本AND公司; SB25-12D超声波清洗机 宁波新芝生物科技股份有限公司;DGX-9143B电热恒温鼓风干燥箱 上海福玛实验设备有限公司;HWS24电热恒温水浴锅 上海一恒科学仪器有限公司;RE-2000B旋转蒸发器 上海亚荣生化仪器厂;SHZ-D(III)循环水式真空泵 巩义市予华仪器有限责任公司。
1.3 方法
1.3.1 麦角固醇标准溶液的配制
精密称取一定质量的麦角固醇和内标物吡嗪,溶于一定体积的氘代氯仿溶液中,混合均匀,取一定体积转入核磁管,待测。
1.3.2 供试品的制备
取食用菌子实体干品适量,研磨,称取0.5 g置于250 mL磨口烧瓶中,加入25 mL质量分数20%的NaOH溶液、25 mL无水乙醇,混匀后85 ℃皂化回流2.5 h,取出冷却、过滤,加入25 mL的石油醚,剧烈振荡15 min,静置1 h,重复操作1 次,合并上层萃取液,水洗2 次,45 ℃旋转蒸发蒸尽石油醚,如此重复5 次实验制备样品,取5 mL氘代氯仿定容。测定前使用0.45 μm滤膜过滤,待测。
1.3.31H-NMR和HPLC检测条件
1H-NMR实验中采用的共振频率为600.18 MHz;脉冲序列zg 30;脉冲宽度9.64 μs;检测温度25 ℃;空扫次数2 次;中心频率2 769.83 Hz,谱宽为δ 20.026 0;接收增益124。
HPLC检测条件:1100液相色谱仪,C18色谱柱(4.6 mm× 250 mm,5 μm);流动相A为甲醇,流动相B为水;洗脱条件:0~2 min,10% A,90% B;2~3 min,100% A,0% B;3~5 min,90% A,10% B。进样量10 μL,流速1.0 mL/min,柱温30 ℃,检测波长270 nm。
1.3.4 核磁共振法定量检测麦角固醇的参数优化
配制一定质量浓度麦角固醇标准品溶液进行弛豫延迟时间、采集时间和扫描次数的考察。设置弛豫延迟时间梯度为5、10、15、20、30、50 s,采集时间梯度为2、3、4、5、6 s,扫描次数为32、64、128、256、320,按设置值从小到大进行核磁实验,以目标特征峰的峰面积值作为参考,保证其稳定不再变化。
1.3.5 麦角固醇标准曲线的绘制
精密称取麦角固醇标准品1.1、2.1、3.1、4、5.3 mg,按由低到高次序分别精密加入1.2、1.2、1.2、0.6、0.6 mg的内标物吡嗪,精准吸取1 mL的氘代氯仿溶液分别溶解,溶解完全后分别准确吸取0.6 mL转入核磁管进行核磁检测,以麦角固醇和内标物质量比作为横坐标,两者的目标特征峰峰面积比作为纵坐标,绘制标准曲线。
1.3.6 定量核磁共振氢谱检测麦角固醇方法学考察
1.3.6.1 检出限和定量限
2020年《中华人民共和国药典》(第四版)规定,核磁共振的检出限和定量限RSN分别为3和10。英国药典规定核磁定量方法的定量限RSN需不小于150。本研究取RSN中较大值作为定量限标准。
实验时精密称取麦角固醇标准品,精确到0.1 mg,氘代氯仿溶液溶解、稀释,配制为不同质量浓度的麦角固醇溶液,按照质量浓度从高到低顺序进行核磁扫描检测,以核磁处理软件自带的RSN计算方法进行麦角固醇目标特征定量峰的RSN计算,分别确定RSN为150和3条件下的麦角固醇质量浓度作为定量限和检出限。
1.3.6.2 精密度实验
称取麦角固醇标准品和内标物吡嗪适量,配制一份麦角固醇和内标物的混合标准溶液,在核磁优化条件下连续扫描5 次。
1.3.6.3 重复性实验
精密称取麦角固醇标准品和内标物吡嗪适量,取氘代氯仿混合溶解完全,配制为6 份相同质量浓度的麦角固醇内标混合溶液,转入核磁管,分别在同等优化条件下扫描。
1.3.6.4 稳定性实验
精密称取麦角固醇和内标物吡嗪适量,取氘代氯仿混合溶解完全,室温放置,在优化后的核磁条件下分别于0.0、6.5、23.5、30.5、48.0 h检测。
1.3.6.5 加标回收实验与计算
精密称取一定质量的麦角固醇标准品和吡嗪内标物,配制为一定质量浓度的麦角固醇与内标标准混合溶液作为加标原溶液;精密称取一定质量的麦角固醇标准品,配制为高质量浓度的麦角固醇加标液,分别取一定体积精密加标3 份原溶液,加标量分别计算标定为1.228 1、2.017 5、2.719 3 mg,混匀后各取0.6 mL核磁检测。按式(1)计算加标回收率:

1.3.7 检测样品中麦角固醇
称取2.5 g食用菌样品粉末,按1.3.2节方法提取麦角固醇,溶于25 mL无水乙醇后取出0.5 mL用于HPLC检测进行对比,其余旋转蒸干后加入5 mL 0.02 mg/mL的吡嗪氘代氯仿溶液溶解,检测样品中麦角固醇含量。
1.3.8 谱图处理
1.3.8.1 谱图的校正、定标与积分
采用MestReNova 9.0软件进行核磁图谱的定标和积分处理。点击MestReNova 9.0软件工具栏的“基线校正”,选择伯恩斯坦多项式拟合方法进行基线校正;选择工具栏的“相位校正”,定位拟调整相位峰,点击鼠标左键调节其相位为零级相位校正;调整特定的峰,点击鼠标右键调节其他远端峰相位为一级相位校正。
完成基线和相位校正后,以氘代氯仿溶剂峰进行定标;点击工具栏的“参考”,选择化学位移δ 7.2峰,标峰时将化学位移更改为δ 7.26,对谱图上其他峰逐个标峰。
积分时放大谱图,取峰信号与基线刚好重叠处为起止点,固定积分区间,内标物吡嗪信号峰积分区间约为δ 8.57~8.63,麦角固醇目标定量峰C-18H积分区间约为δ 0.60~0.66,确保积分的重复性良好。图谱处理时手动进行相位校正和基线校正,对内标物和样品的定量峰分别进行5 次积分,取其平均值,得到积分结果。
1.3.8.2 多重峰分析
选择MestReNova 9.0核磁处理软件进行多重峰分析,批量处理已标定、积分完毕的峰,得到包含峰化学位移、峰等级(单/双峰/多重峰)和氢质子数等信息的谱图分析结果。
1.3.8.3 RSN的计算
采用MestReNova 9核磁处理软件,点击谱图,基线与相位校正后,对拟处理目标峰进行化学位移标定,在谱图上选择一段平滑的基线区域计算噪声区,根据需要的信号范围,采用软件工具栏中SNR Peak Calculator自动计算出标定的目标峰RSN。
1.3.8.4 线性拟合
采用Topspin 3.6.2软件工具栏中“Start Fit”进行核磁的线性拟合,将拟合后的谱图在MestReNova 9.0中进行常规积分,获得去除杂质峰干扰后的样品定量检测峰积分结果。
1.3.9 计算公式
定量核磁的基本原理是图谱中信号响应值(即峰面积WX)与待测物(如质子,NX)数目呈正比,而与待测物的化学性质无关,一般只要对该化合物中某一基团上质子引起的峰面积进行比较,即可求出其绝对含量。对目标信号积分,按照1H-NMR内标法,依据式(2)定量,计算食用菌中麦角固醇的含量:
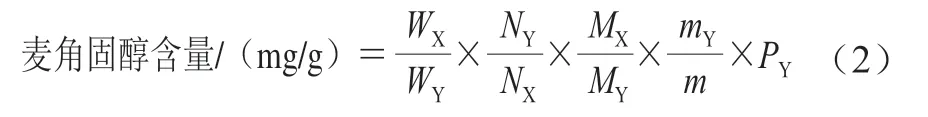
式中:MX和MY分别为待测物和内标物的相对分子质量;m为测定混合物样品的质量/mg;mY和PY分别为内标物的质量/mg和纯度;NX/NY和WX/WY分别为图谱中待测物目标峰及内标对应核的个数及峰面积。
HPLC检测的麦角固醇含量按式(3)计算:
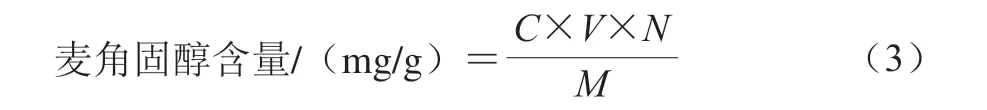
式中:C为麦角固醇质量浓度/(mg/mL);V为样品定容体积/mL;N为稀释倍数;M为萃取时样品质量/g。
1.4 数据处理与统计分析
采用Origin 8、Excel进行数据的统计、绘图分析。
2 结果与分析
2.1 1H-NMR氘代溶剂和内标物的选择
氘代氯仿试剂在核磁共振波谱分析中起溶解样品和信号锁场的双重作用,氘代氯仿试剂必须对样品和内标物溶解性都非常好,不会导致谱图信号重叠。麦角固醇作为小分子物质,相对分子质量为396.65,均可溶于乙醇、乙醚和氯仿等有机溶剂,几乎不溶于水,其中在氘代氯仿中两者溶解度均较大,麦角固醇达到32.258 1 g/L, 且氘代氯仿较经济易得,因此选择氘代氯仿作为溶剂。吡嗪(对二氮杂苯)在其2、3、5、6位上含有4 个等价氢,相对分子质量为88.088,易溶于乙醇、乙醚、氯仿和水,是核磁定量实验中常见的内标物。称取麦角固醇、吡嗪,以氘代氯仿充分溶解,取0.6 mL核磁检测,以溶剂对照,结果如图1所示。
如图1A所示,氘代氯仿溶剂峰在δ7.26处出现,四甲基硅烷在δ0.0峰,在核磁共振波谱的1H、13C和29Si谱中起到标定化学位移零点的作用;根据内标物质在选定氘代氯仿溶剂中的溶解性和MestReNova软件模拟内标物和待测样品的核磁氢谱,内标物吡嗪在出峰位与待测样品的谱峰没有重叠,与定量峰的间距也较为合理,图1B中,麦角固醇的信号峰在δ0~5.6之间,与氘代氯仿峰(δ7.26)互不干扰可选作溶剂。由图1C对比显示,内标物吡嗪在δ8.60处出峰,吡嗪对目标物麦角固醇的峰信号互不产生干扰,且不干扰样品的定量峰,在氘代氯仿中能较好地溶解,可选吡嗪为内标物进行定量实验。
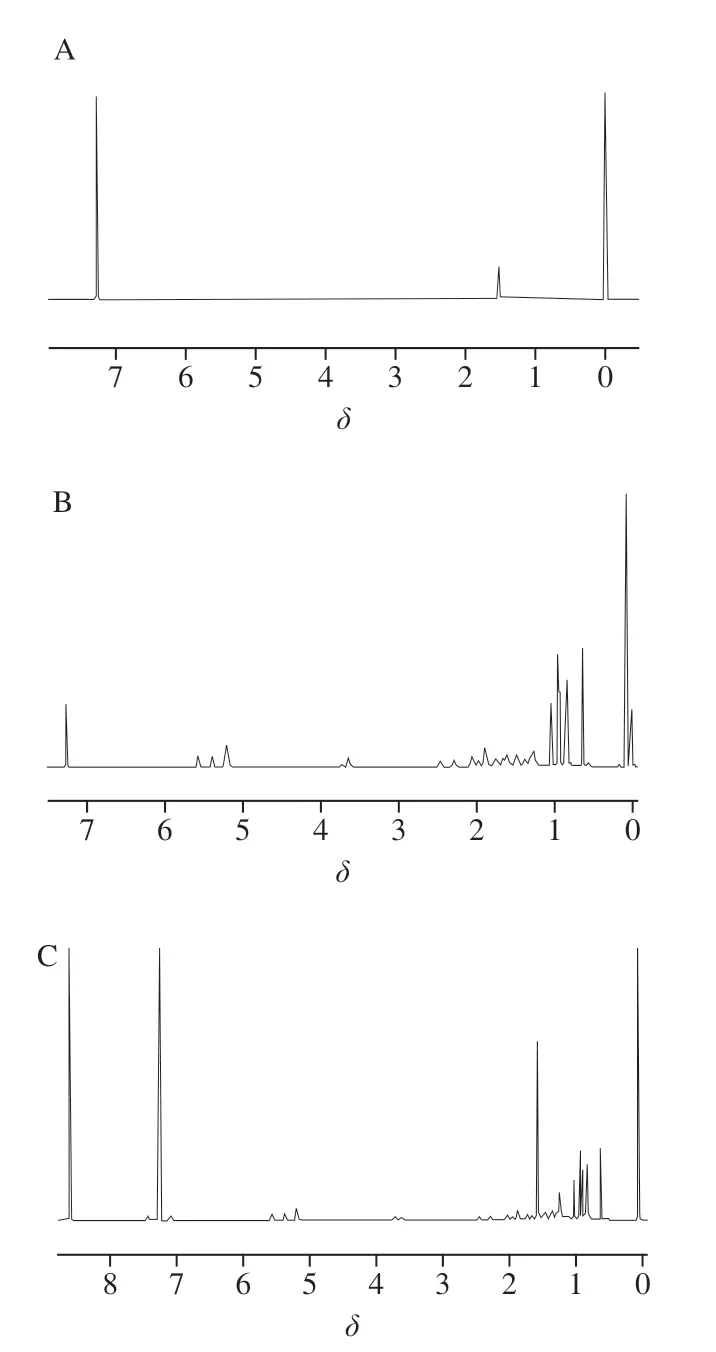
图1 氘代氯仿(A)、麦角固醇标准品(B)、麦角固醇标准品与 内标物吡嗪的混合(C)图谱Fig. 1 NMR spectra of deuterated chloroform (solvent blank) (A), ergosterol standard (B), and a mixture of ergosterol and pyrazine standard (C)
2.2 核磁检测食用菌麦角固醇检测体系的建立
2.2.1 麦角固醇分子结构信号峰归属
以氘代氯仿作为溶剂,配制麦角固醇的溶液,于600 MHz核磁仪器上检测,其质子信号峰归属情况 见表1、图2。
少女将宽大的滑翔翼丢在地上。下落时狂烈的山风,吹落了她束缚头发的皮绳,她那深栗色的卷发,此刻正杂乱地披散在肩上,随着风轻轻鼓荡,像狂野的浪。大大的眼睛黑而深邃,浅棕色的皮肤,精瘦紧俏的脸颊,饱满紧致的双唇,透露着一种野性的美感。她怒气冲冲地走过来,劈头盖脸地骂道:“你这个笨蛋!你不要命了吗!”
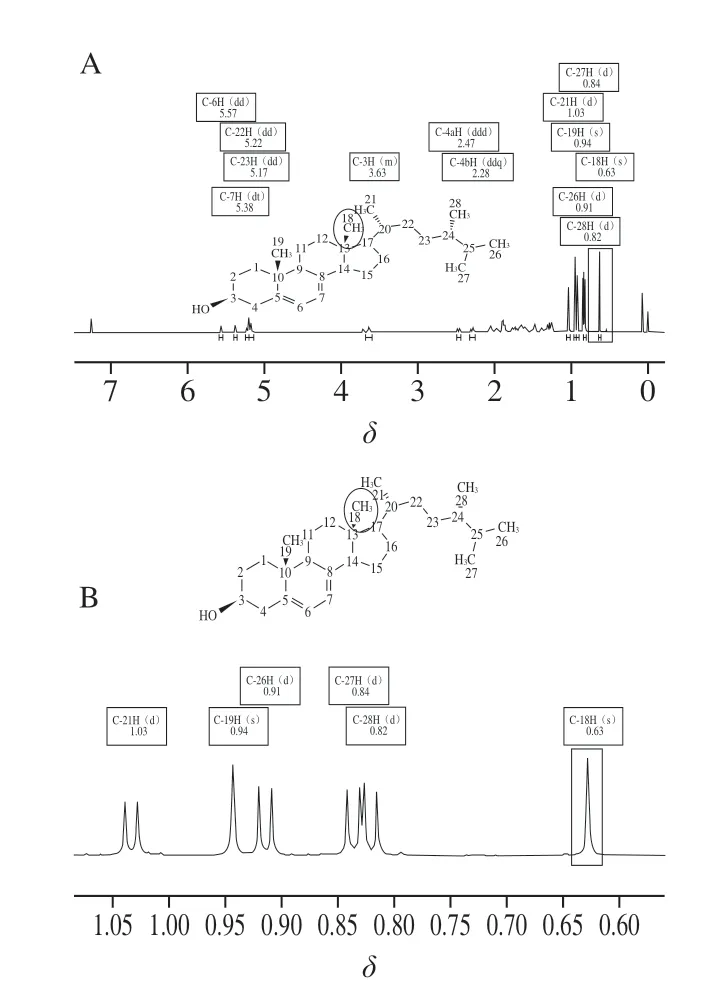
图2 麦角固醇质子信号峰归属Fig. 2 Assignment of ergosterol proton signal peaks
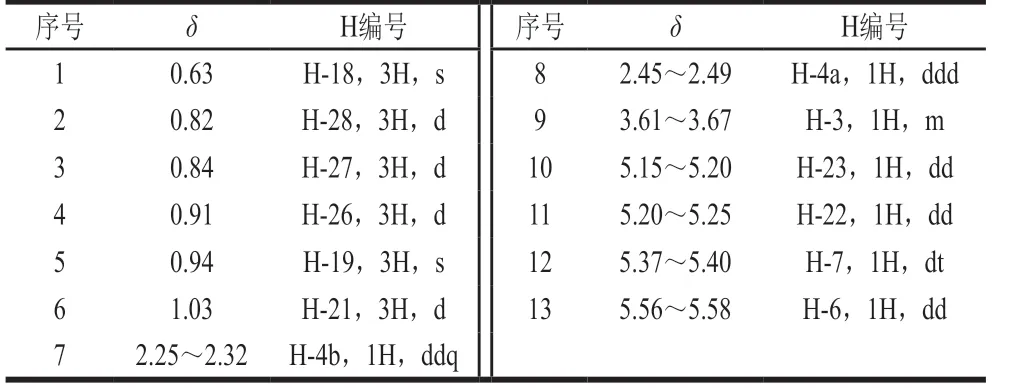
表1 麦角固醇的质子信号峰归属Table 1 Assignment of ergosterol proton signal peaks
从表1可知,在化学位移δ0.63处C-18H与δ0.94处 C-19H为单峰外,其余质子峰均为双峰或混峰,对于待测物及内标,均应尽量选择含多个原子的孤立尖锐单峰作为定量峰,以保证有足够的积分范围。
2.2.2 食用菌麦角固醇目标定量峰的确定
采用1H-NMR进行定量时,应对被测样品中的各个质子信号进行归属,明确每个信号峰所对应的质子数,选择合适的信号峰作为定量特征峰[28]。配制不同质量浓度麦角固醇与内标物吡嗪的标准混合溶液,常规检测条件下检测,以内标定量法进行核磁检测结果计算,与理论质量进行对比,观察质子峰峰面积与麦角固醇上样量的相关性。计算结果如表2所示。
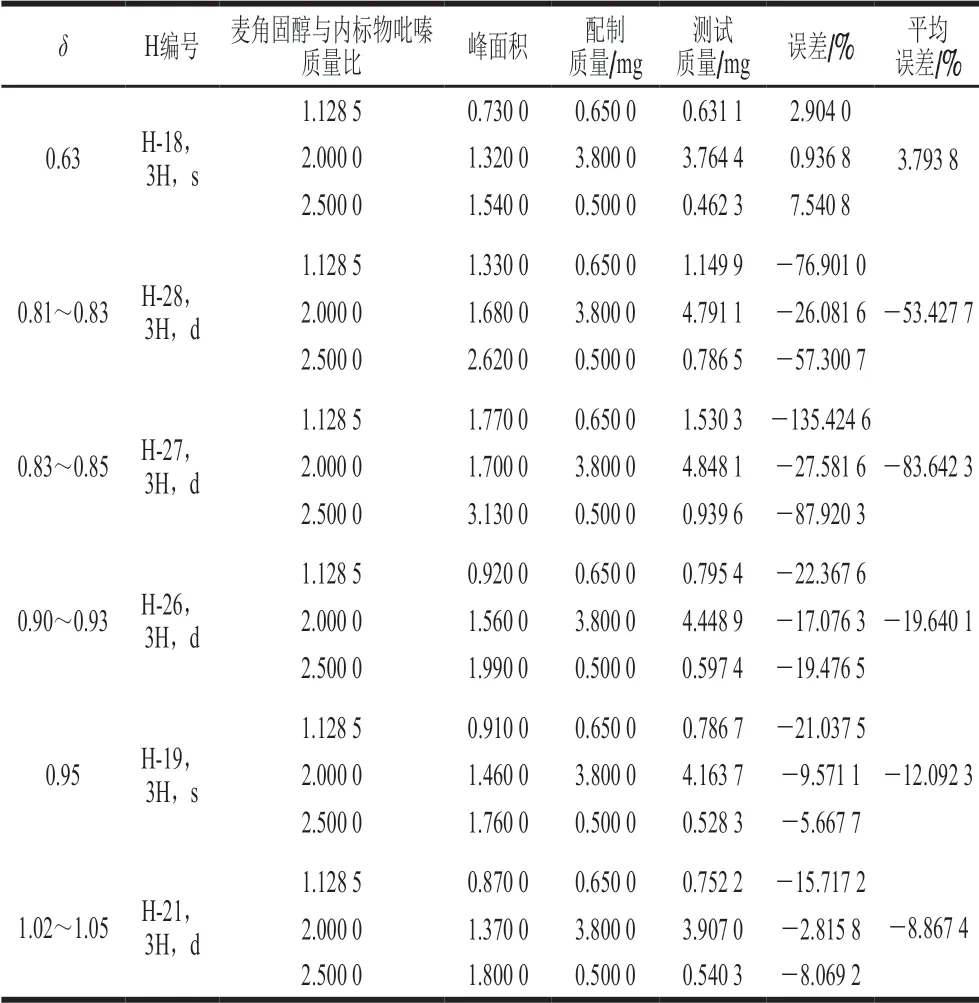
表2 麦角固醇的质子信号峰定量验证 Table 2 Quantitative verification of proton signal peaks of ergosterol
表2结果显示,δ0.63质子单峰相比δ0.95和δ1.02~1.05处单峰和双峰的计算结果与理论值误差更小。定量质子特征峰应基线稳定能实现基线分离,如图3所示,谱图中能实现基线分离的质子信号峰分别仅有处于高场区δ0.63(单峰)和δ1.04(双峰),尽管δ0.95处的质子单峰在一定条件下检测积分相对稳定,但受右侧2 个双峰影响,信号峰基线不稳未能实现基线分离,无法作为定量特征峰。
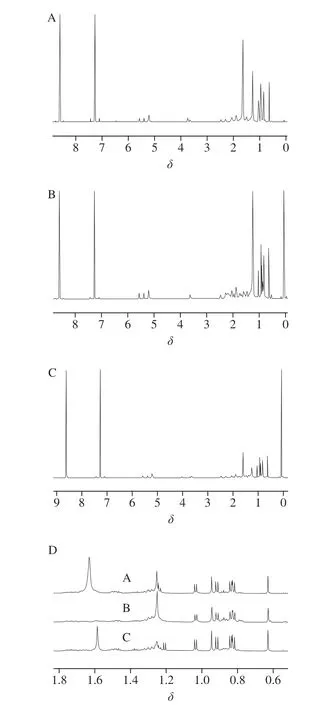
图3 核磁检测鸡油菌(A)、茶树菇(B)样品和麦角固醇 标准品(C)谱图及局部图(D)Fig. 3 NMR full spectra of chanterelle (A), Agrocybe aegerita (B) and ergosterol standard (C) and partial enlarged spectra (D)
通过麦角固醇标准品和食用菌样品谱图的放大对比,食用菌样品中存在的复杂基质均对δ0.95和δ1.04的信号峰产生不同程度的干扰,均无法成为定量目标峰,如图3所示。
核磁定量中优先选择单峰作为定量峰,放大δ0.63的单峰观察,干扰δ0.63单峰,主要是在其右侧形成一个信号相对较弱且不能与主峰基线分离的小峰,如图4b所示,采用核磁谱图处理软件(Bruker TopSpin软件)去卷积处理方法,可对目标峰进行线性拟合,拟合后单独积分,进而排除杂质峰对主峰的积分干扰,最终确定选择C18甲基质子单峰C-18H(s)为定量目标峰,通过峰面积积分对样品中麦角固醇进行定量。
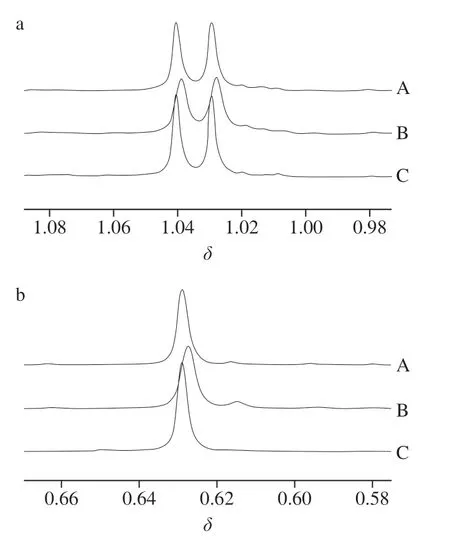
图4 化学位移δ0.63单峰与δ 1.04双峰处信号峰放大图谱Fig. 4 Spectral amplification of signal peak at δ 0.63 and double peaks at δ 1.04
2.3 1H-NMR检测麦角固醇条件的优化
2.3.1 弛豫延迟时间
弛豫过程分为自旋-晶格弛豫(纵向弛豫)和自旋-自旋弛豫(横向弛豫),自旋-晶格弛豫是指处于高能态的核自旋体系将能量传递给周围环境(晶格或溶剂),自身回到低能态的过程,所需时间为自旋-晶格弛豫时间,也称纵向弛豫时间T1,纵向弛豫反映了自旋体系与环境之间的能量交换。通常弛豫时间的长短对核磁定量分析很重要,是定量核磁中非常重要的一个参数,弛豫时间较短会影响定量核磁的精密度和准确度,足够的弛豫时间能确保已激发的原子有充足时间恢复到基态并再次激发,从而使信号多次采集并积累,提高灵敏度。
核磁实验中确定弛豫延迟时间,通过设置不同弛豫延迟时间梯度,按时间从小到大进行实验,对比峰面积值,当峰面积值基本不再变化时,表明达到弛豫延迟时间不小于5Tl要求。研究中采用这个方法进行弛豫延迟时间的确定[30]。配制麦角固醇标准样品,扫描次数128,采样时间4 s,其他条件确定下进行弛豫延迟时间的优化,麦角固醇与内标物峰面积比值的结果如表3所示。
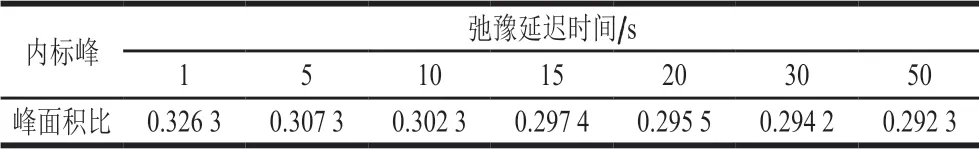
表3 弛豫延迟时间的优化Table 3 Optimization of relaxation delay time
如表3所示,固定积分范围积分,同一份样品分别在不同弛豫时间下扫描,随着延迟时间的延长,峰面积比逐渐下降,到15 s之后,下降趋势减缓稳定,选择15 s作为最佳的弛豫延迟时间。
2.3.2 采集时间
采集时间与谱图的数字分辨率有很大的关系,两者互为倒数关系,即采集时间越长,则分辨率越小,采集时间选择范围一般为2~4 s[31],自由感应衰减(free induction decay,FID)信号经过傅里叶变换才能转换成可看谱图,如果采集时间小于信号完全衰减成噪音的时间,FID信号将被截尾,截尾作用会把假信号带进谱图,使谱图发生畸变。若采集时间大于完全衰减成噪音的时间,则会因为采集到更多的噪音而降低谱图的分辨率。探讨与优化合适的采集时间,保证FID信号衰减完全,提高仪器的使用效率。
在优化的弛豫延迟时间15 s条件下,设置扫描次数为128 次,时间梯度为2、3、4、5、6 s,优化采样时间,结果如表4所示。
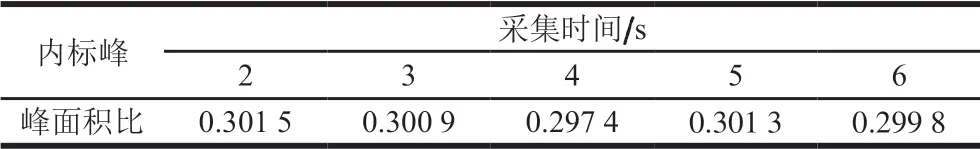
表4 采集时间的优化 Table 4 Optimization of acquisition time
在2~6 s的采集时间中,随着采集时间的延长,峰面积比值在0.3、0.2处稳定波动,为节约实验时间,后续实验选择2 s作为最优采集时间。
2.3.3 扫描次数
核磁定量实验中,为获得高信噪比的谱图,一般通过加大样品的上样量或在样品量较少时增加扫描次数获得。在上样量一定时,较少的扫描时间内,数据的重复性较差,增加扫描次数,数据重复性较好,谱图信噪比高,得到平滑的谱图,但会延长占机时间,需要确定适合的扫描次数。设置采集时间2 s、弛豫延迟时间15 s,设扫描次数分别为32、64、128、256、320,优化扫描次数,结果如表5所示。
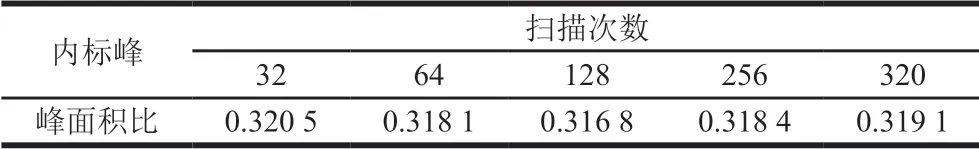
表5 扫描次数的优化 Table 5 Optimization of number of scans
由表5可知,峰面积比值首先随着扫描次数的增加而下降,此时数据的重复性较差,到256 次时缓慢增加并基本趋于稳定,此时数据较稳定,重复性好。考虑为获得更高的信噪比,故选择256 次作为最佳的扫描次数。
2.4 1H-qNMR检测麦角固醇质量浓度线性比例关系
配制不同质量浓度的麦角固醇和吡嗪混合溶液,在优化后的条件下核磁检测。设置麦角固醇质量为Wx,吡嗪质量为WIS,麦角固醇峰面积为Ax,内标物吡嗪峰面积为AIS。以麦角固醇标准品和内标物的质量比作为横坐标,麦角固醇标准品和内标物的峰面积比作为纵坐标,绘制标准曲线。麦角固醇与内标物吡嗪质量比为0.55~5.30 mg/mg范围内,线性回归方程为Y=0.130 6X+0.008 3,相关系数R2大于0.999 7,表明线性关系良好,能满足核磁定量需要。
2.5 1H-qNMR检测麦角固醇方法的评价
2.5.1 检出限和定量限结果
样品测定时,根据中国药典和英国药典对qNMR要求(RSN≥150)。在一定硬件条件下,样品浓度无法改变时,可以通过增加或累积扫描次数增加信号强度从而提高RSN。但是扫描次数并不是越多越好,太多的扫描次数无疑延长了测定时间。以核磁处理软件自带的RSN计算方法进行麦角固醇目标特征定量峰的RSN计算,分别确定RSN为150与3条件下的麦角固醇质量作为定量限和检出限,结果如表6所示。
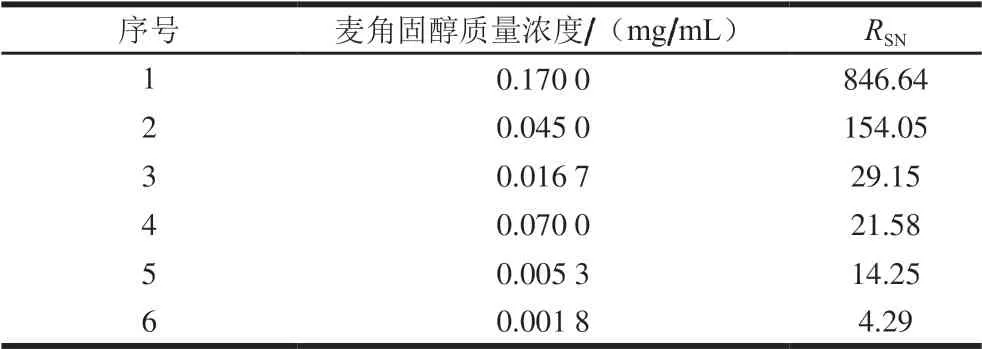
表6 核磁麦角固醇质量与信噪比Table 6 Ergosterol concentration and corresponding signal to noise ratio
如表6所示,在0.6 mL的氘代溶液中,当麦角固醇质量浓度为0.001 8 mg/mL时,RSN为4.29,确定为检出限;当麦角固醇质量浓度为0.005 3 mg/mL时,RSN为14.25,可作为其定量限。
根据英国药典规定,核磁氢谱定量方法若达到精确定量,RSN应不小于150,在麦角固醇质量浓度为0.045 0 mg/mL时,RSN为154.05,故为保证核磁定量精确度,确定最终定量限为0.045 0 mg/mL。
2.5.2 方法精密度、重复性与稳定性分析
在核磁优化实验条件下,日内扫描5 次分析仪器的精密度;精密称取麦角固醇和吡嗪标准品配制质量浓度相等的6 份溶液,并在0.0、6.5、23.5、31.0、48.0 h条件下核磁检测,结果如表7所示。
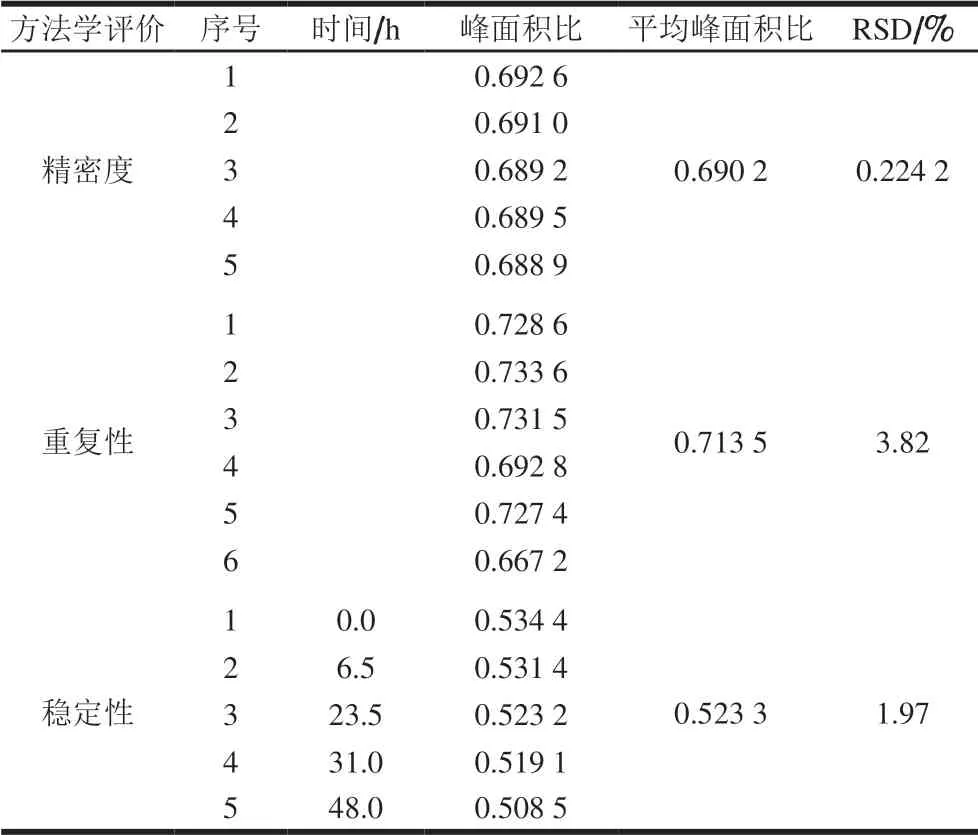
表7 仪器精密度、重复性与稳定性检测Table 7 Evaluation of instrumental precision, repeatability and stability
从表7可知,日内扫描5 次的峰面积比相对标准偏差(relative standard deviation,RSD)为0.224 2%,6 份重复样品的检测结果RSD为3.82%,以峰面积法定量时,0.0~6.5 h内峰面积比变化较小,RSD为0.392 1%;0.0~23.5 h内,RSD为1.550 6%;0~48 h内的RSD为1.97%,样品室温放置48 h略有下降后趋于稳定,这表明在核磁优化实验条件下,仪器的精密度与方法稳定性良好,使用峰面积法核磁定量检测能满足定量需求。
2.5.3 加标回收实验
通过回收率实验准确地衡量测定值与真实值的接近程度。如表8所示,以标准品加标,回收率在95.246 6%~109.054 6%之间,其RSD为5.495 2%。表明分析方法与测量系统准确度良好。
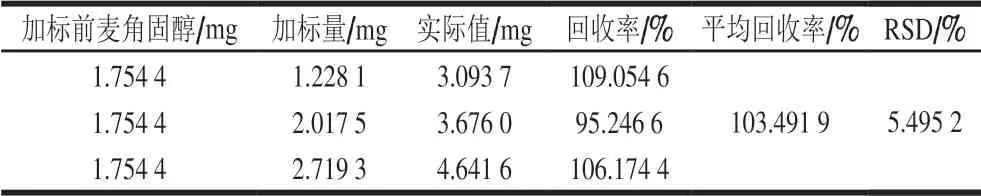
表8 加标回收实验结果Table 8 Recoveries for spiked standards
2.6 样品检测与方法专属性
将已制备待测样品溶液溶于25 mL的无水乙醇,取0.5 mL用于HPLC检测;剩余样品经真空旋转蒸发去除乙醇,溶于5 mL的氘代氯仿溶液中,取1 mL到核磁管中,加入精密配制质量浓度为1.2 mg/mL的内标物吡嗪溶液0.5 mL,在优化的核磁条件下检测,分别采用核磁软件MestRenova 9和Bruker Topspin3.6.2处理数据与谱图,结果如图5与表9所示。
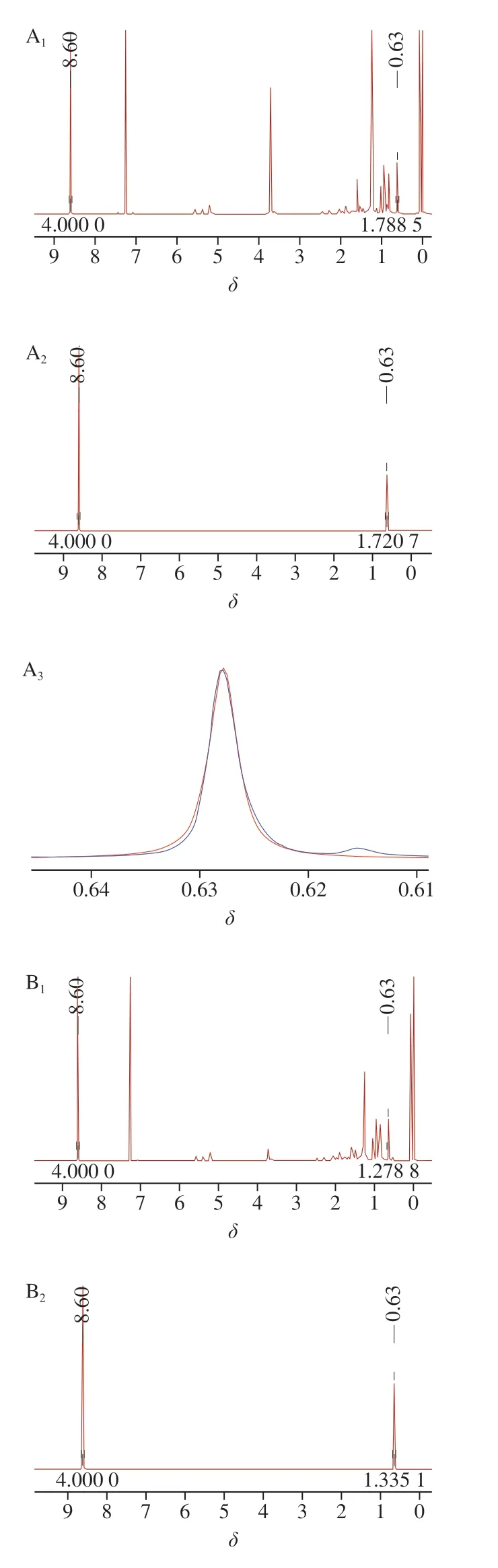
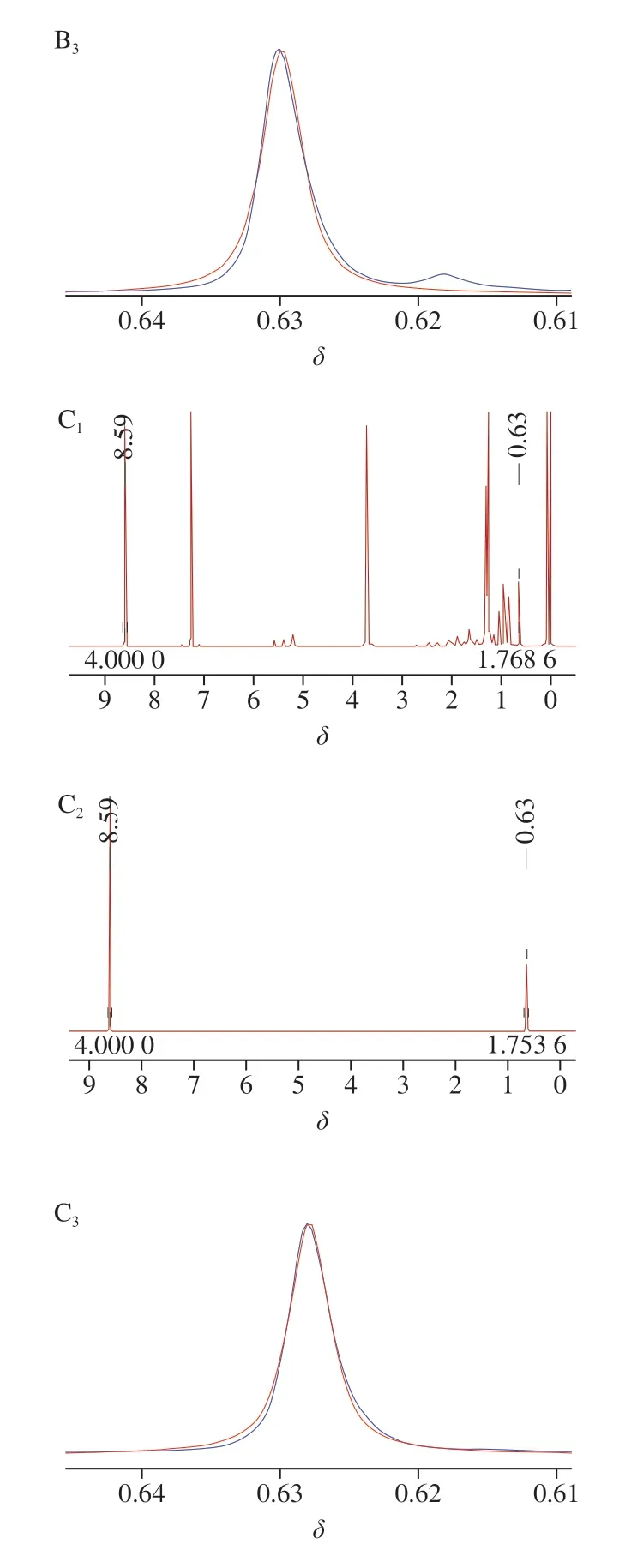
图5 鸡油菌(A)、茶树菇(B)与虫草(C)样品麦角固醇 定量峰去卷积拟合谱图Fig. 5 Deconvoluted and curve-fitted spectra of quantitative ergosterol peaks of chanterelle (A), Agrocybe aegerita (B) and Cordyceps (C)
定量核磁研究中面积积分值的准确性对定量结果有很大的影响。使用直接积分时,2 个相邻峰的谱图重叠以及基线都会对峰面积造成影响引起结果的偏差,可选择直接普通积分和去卷积积分2 种积分方式对得到的氢谱谱图进行对比,能准确获取该化学位移处峰的面积信息。核磁谱图中有峰重叠或受噪声影响过大,当对谱图直接进行积分(中切法)时,得到的面积有一定偏差,如表9所示。可使用去卷积方法算出各峰的峰面积,将得到的谱图用Bruker Topspin软件,对需要去卷积的目标峰部分进行去卷积拟合积分,得到相应的峰面积,如图5所示。
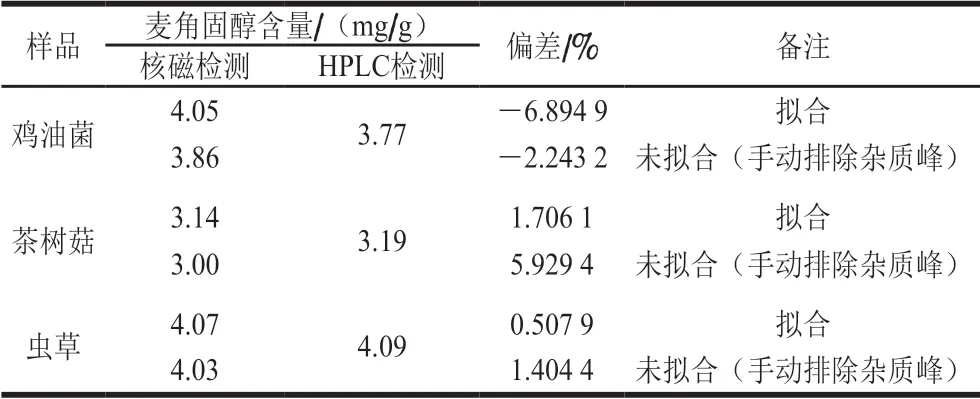
表9 食用菌样品核磁定量检测结果(n =3)Table 9 Results of NMR for ergosterol in several edible fungi samples(n = 3)
核磁谱图中峰的形状主要是由洛伦兹(Lorenz)方程给出,实际检测过程中,由于场的不均匀性和加权函数,会产生部分高斯(Gauss)线形状,若单纯采用洛伦兹或纯高斯进行去卷积拟合时,2 种峰去卷积拟合残差较大,计算出的结果区别较大,在适当情况下,多数对称的峰均可调节高斯和洛伦兹线性比例以近似代替[32],采用洛伦兹和高斯以一定比例进行去卷积拟合时发现,当洛伦兹比高斯为4∶1时,拟合曲线与原来的峰形最相似,且与HPLC测定的麦角固醇结果接近。从表9可知,经拟合处理后积分,检测结果较靠近HPLC检测值,说明在样品检测以后进行去卷积拟合处理,能得到更精确的定量值,这表明在样品核磁检测中,当目标定量峰周围存在杂峰干扰时,无法精确积分得到较为准确的定量结果,可通过洛伦兹和高斯线性比例调节进行去卷积分析,能排除杂峰的干扰,提高定量准确性。
3 结 论
根据麦角固醇核磁谱图质子信号峰归属,以氘代氯仿作为溶剂与吡嗪作为内标物,通过优化核磁检测参数建立1H-NMR检测食用菌中麦角固醇的方法并进行方法学考察,通过食用菌样品实证检测,论证检测方法专属性与可行性。研究结果表明,麦角固醇核磁谱图中C-18H化学位移δ0.63处的甲基质子单峰可作为麦角固醇的目标定量峰,以采集时间2 s、弛豫延迟时间15 s和扫描次数256 次作为优化核磁检测条件,麦角固醇与内标吡嗪的质量比与峰面积之比呈良好的线性关系,可获得稳定的面积比值积分值,在0.55~5.30 mg/mg范围内,线性回归方程为Y=0.130 6X+0.008 3,相关系数R2大于0.999 7;经方法学评价,方法检出限(RSN≥3)与定量限(RSN≥150)分别为0.001 8 mg/mL与0.045 0 mg/mL,方法的精密度与稳定性RSD均小于2%;采用洛伦兹与高斯比例为4∶1时进行去卷积拟合时发现,拟合曲线与原来的峰形最相似,并与HPLC法测定的麦角固醇结果相近,说明在样品检测后进行去卷积拟合处理,能得到更精确的定量值;3 种食用菌实际样品检测验证该方法的专属性与可行性,表明建立的方法对于食用菌中麦角固醇定量检测具有快速准确的优势。
