超高效液相色谱-串联质谱法测定虾肉中52种兽药残留
2021-11-04吴益春陈璐郭海波罗海军王晓煜周勇
吴益春 ,陈璐,郭海波*,罗海军,王晓煜,周勇
1.舟山市食品药品检验检测研究院(舟山 316013);2.宁波大学食品与药学学院(宁波 315800);3.浙江海洋大学食品与药学学院(舟山 316022)
近年来,随着人们对水产品需求的不断扩大,为增加养殖产量、提高经济效益,各类兽药被广泛应用于水产养殖生产。由于兽药在使用过程中存在滥用、误用及不遵守休药期等现象,水产品中存在药物残留的现象较为普遍。这些有兽药残留的水产品,经食用后在人体中蓄积,可能会产生致畸、致癌的严重危害。测定兽药残留及其代谢物的检测方法有气相色谱-质谱法[1-2]、高效液相色谱法[3]、高效液相-串联质谱法[4-8]、超高效液相色谱-四极杆-飞行时间质谱法[9-10]、三重四极杆复合线性离子阱质谱法[11-12]、超高效液相色谱-四极杆/静电场轨道阱高分辨质谱法[13-14]等,由于不同兽药的理化性质存在较大差异,且同一种兽药还有同系物、异构体、降解产物及共轭物的存在,兽药残留检测主要以分类检测为主[9],尚无分析方法可以同时涵盖所有的药物种类。试验针对虾肉中的大环内酯类、酰胺醇类、硝基咪唑类、磺胺类、四环素类、喹诺酮类等6个大类52种兽药残留,开发QuEChERS EMR-Lipid结合超高效液相色谱-串联质谱法,为大批量虾类兽药残留的筛查分析提供参考。
1 材料与方法
1.1 材料与仪器
新鲜淡水虾、海水虾(各40批,随机采集于浙江舟山市菜市场、农贸市场、超市);乙腈、甲酸(色谱纯,德国Merck公司);去离子水Sartourius系统纯净水、十八烷基键合硅胶(C18)、N-丙基乙二胺吸附剂(PSA)、0.22 μm有机针式滤膜、增强去脂萃取盐包(m无水MgSO4∶mNaCl=4∶1)、增强型去脂分散净化管(EMR-Lipid dSPE管,1 g,15 mL):上海安谱科技股份有限公司;红霉素等52种兽药(纯度均≥90.0%,标准品,德国Dr.Ehrenstorfer公司)。
1290-6470超高效液相色谱-质谱联用仪(美国安捷伦公司);5804R冷冻离心机(德国艾本德公司);MS-3涡旋振荡器(德国IKA公司);UV-R超纯水机(法国Millipore公司);MS205DU电子天平(瑞士Mettler-Toledo公司);KQ-6000DV数控超声波清洗器(昆山市超声仪器有限公司);N-EVAPTM111氮吹仪(美国Organomation公司)。
1.2 色谱-质谱条件
液相色谱柱ZORBAX Eclipse plus C18柱(3.0 mm×150 mm,1.8 μm);柱温30 ℃;进样量5 μL;流动相A为0.2%甲酸水溶液、流动相B为乙腈溶液(含0.2%甲酸);流速0.5 mL/min。梯度洗脱程序:0~0.5 min,2% B;0.5~1.8 min,2%~15% B;1.8~3.5 min,15%~20% B;3.5~6.0 min,20%~25% B;6.0~7.0 min,25%~30% B;7.0~11.0 min,30%~35% B;11.0~16.0 min,35%~100% B;16.0~26.0 min,100% B。
离子源,电喷雾离子源;扫描方式,正负离子切换扫描;检测方式,多反应监测(MRM)模式检测;毛细管电压正离子4500 V,负离子3500 V;干燥气温度350 ℃;干燥气流量5 L/min;雾化气压力45 psi;鞘气温度300 ℃;鞘气流速11 L/min;喷嘴电压500 V,其他质谱条件见表1。

表1 代表性化合物质谱参数
1.3 标准溶液的配制
准确称取适量标准品,置于100 mL棕色容量瓶中,用乙腈分别溶解并定容至刻度,配制成100 mg/L的标准储备溶液,在-18 ℃下避光保存备用。混合标准溶液:分别取上述标准储备溶液,用乙腈配制成1 mg/L的混合标准溶液,在2~4 ℃冰箱保存。
配制基质匹配混合标准溶液。取若干空白试样,按1.4的方法处理与净化,用该空白试样溶液和混合标准溶液配制成质量浓度2~500 ng/mL基质匹配的混合标准系列溶液。该系列溶液临用现配。
1.4 样品前处理
准确称取2.0 g(精确至0.01 g)均质后的试样置于50 mL聚丙烯离心管中,加入2 mL超纯水,加盖旋涡混匀1 min后,加入8 mL 1%甲酸乙腈,加入增强去脂萃取盐包,涡漩混匀1 min,超声提取30 min,按6000 r/min离心5 min,取上清液,待净化。取2 mL上清液移入QuEChERS EMR-Lipid dSPE管中,涡漩混匀1 min,静置5 min,按6000 r/min离心5 min后,将上清液移入15 mL离心管中,于50 ℃水浴氮气吹干,加入1 mL 20%乙腈溶液复溶,旋涡混匀30 s,经0.22 μm滤膜过滤后,供UPLC-MS/MS测定。
2 结果与分析
2.1 色谱条件的优化
优化碎裂电压、碰撞能量等仪器条件设置,筛选高响应特异离子对等质谱条件,采用正负离子扫描多反应监测模式,一针进样完成所有目标物的检测。各代表性化合物质谱参数见表1,6种代表化合物的MRM色谱图见图1。
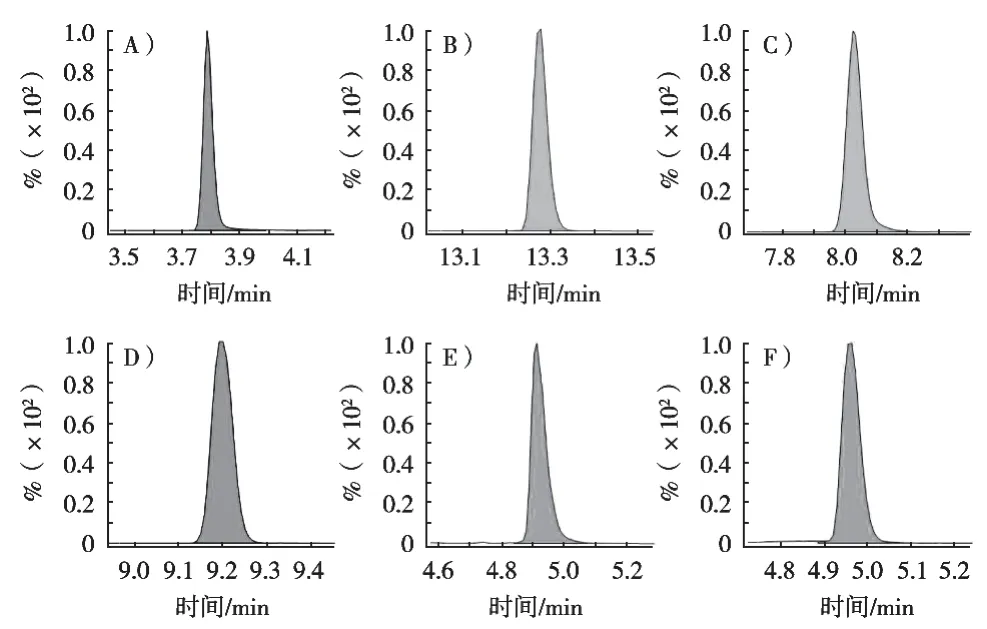
图1 每类兽药代表性化合物在多反应监测模式下的色谱图
2.2 样品前处理条件优化
2.2.1 提取条件优化
选择南美白对虾为试验对象,通过添加回收试验,考察甲醇、乙腈、乙酸乙酯、丙酮4种试剂的提取效率。试验结果表明,乙酸乙酯和丙酮对虾肉中的杂质萃取较多,影响兽药残留回收率,平均回收率均低于50%。选取甲醇和乙腈2种有机溶剂进行考察,结果显示乙腈对多数目标物均有较好的溶解性和较高的提取率[15-16],各种兽药平均回收率为89.4%,优于甲醇。因此,试验选择乙腈作为提取溶剂。
2.2.2 净化条件优化
由于虾肉中干扰物主要是脂肪、蛋白质、色素等,为有效去除干扰物质,考察常用净化材料C18、PSA、增强型去脂分散净化管对于待测目标物含量的影响。结果表明,PSA对样品中一些强极性杂质、脂肪酸、色素等具有良好的净化效果,但PSA吸附磺胺类药物,磺胺类药物回收率最低,仅34.0%,C18粉对样品中蛋白质和脂类物质有一定吸附效果[17],但净化效果不够理想,个别样品存在净化后溶液较为浑浊现象。增强型去脂分散净化材料能减少脂质基质对兽药的吸附干扰,各种兽药残留最低回收率能够达到60%以上,而且净化后的液体不存在浑浊现象。因此,最终选择增强型去脂分散净化管作为净化剂。
2.3 线性范围、检出限与定量限
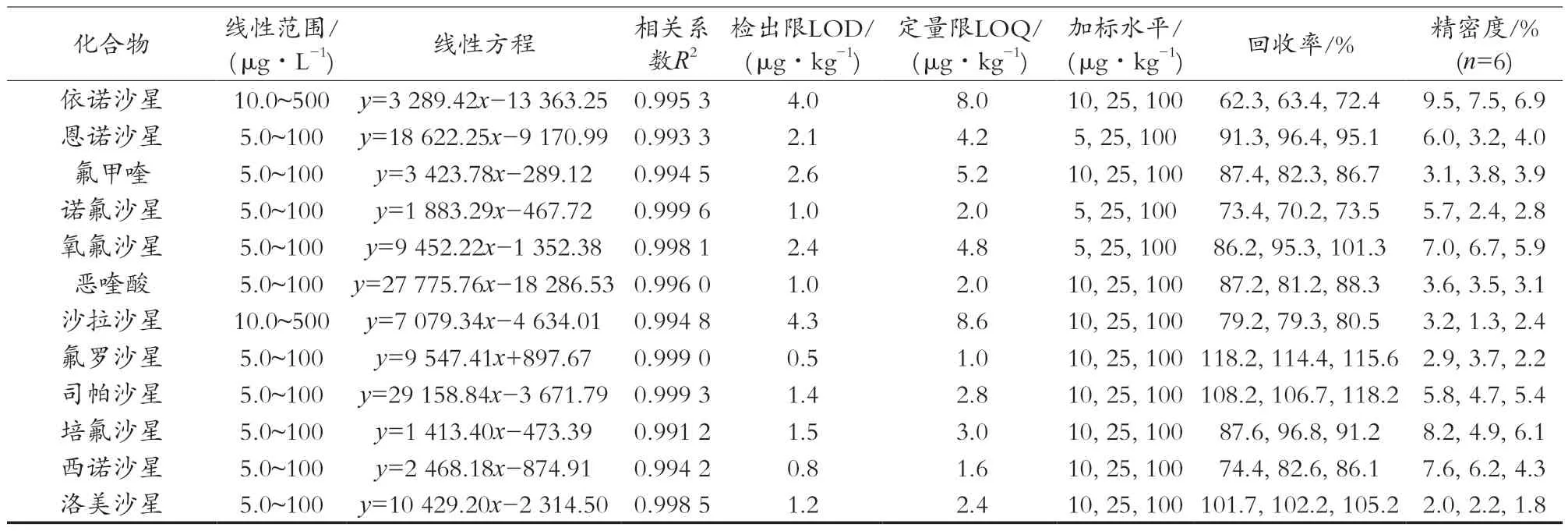
采用基质匹配标准曲线进行定量,以抵消基质效应的影响,以目标物的峰面积为纵坐标(y),以相应的质量浓度质量(μg/L)为横坐标(x)绘制标准曲线。试验结果表明,各目标物线性关系良好,相关系数均大于0.99,线性范围较宽。以3倍信噪比(S/N)计算检出限,各种兽药检出为0.36~14.00 μg/kg,结果见表2。
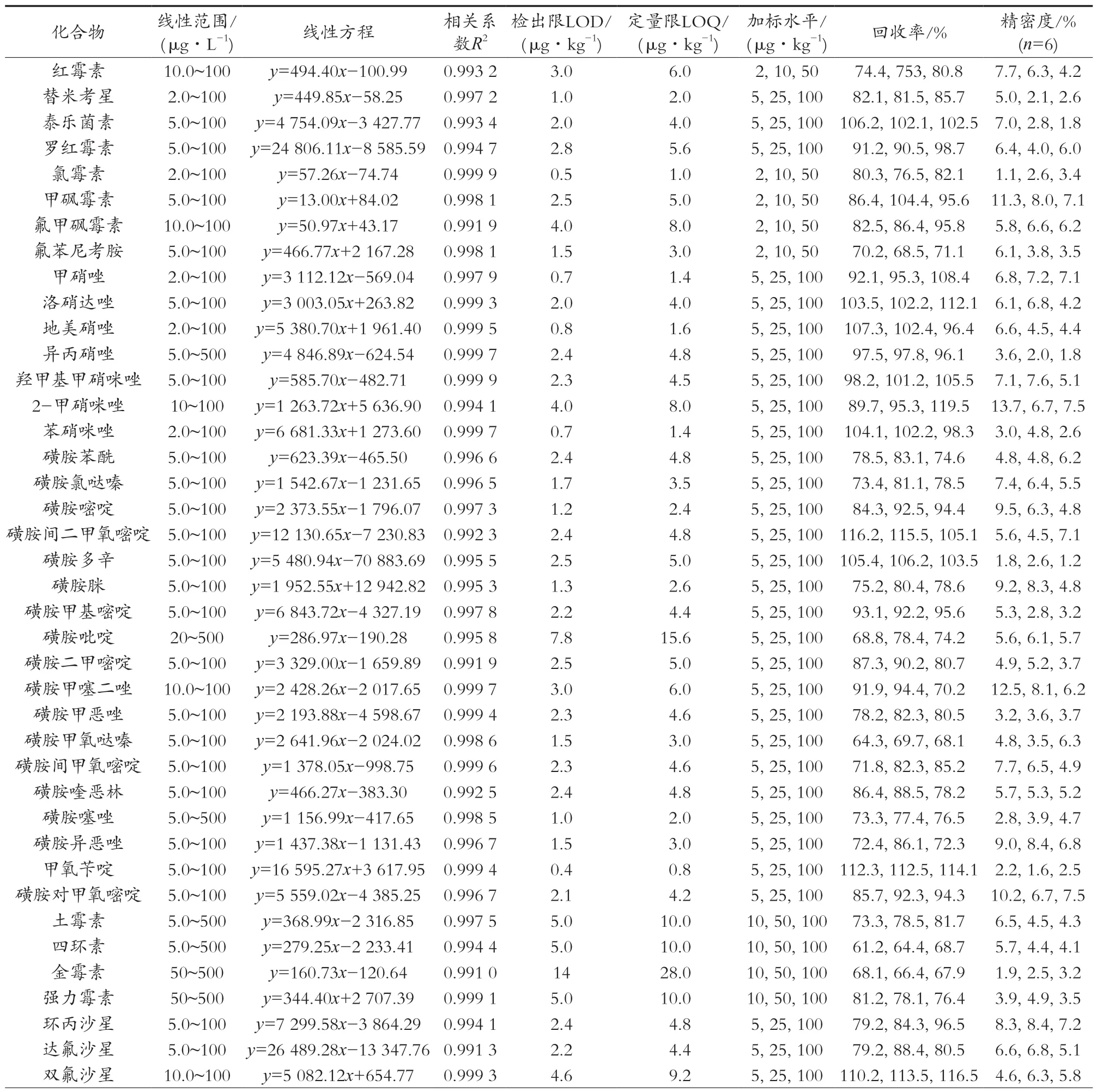
表2 52种兽药的线性范围、线性方程、相关系数、检出限、定量限、加标回收率和精密度
2.4 方法回收率与精密度
在空白南美白对虾中分别添加低、中、高3个水平的混合标准溶液,每个添加水平重复测定6次,同时计算相对标准偏差来考察精密度,具体结果见表2。结果表明,3个添加水平下目标化合物的平均回收率为61.2%~119.5%,相对标准偏差为1.0%~13.7%,符合实际检测要求。
3 结论
该试验建立了虾肉中大环内酯类、酰胺醇类、硝基咪唑类、磺胺类、四环素类、喹诺酮类6个大类52种理化性质相差较大目标化合物的分析检测方法。为获得满意的分析物回收率,对仪器条件、提取方式、净化方式进行优化,52种兽药检出限为0.36~14.00 μg/kg,回收率为61.2%~119.5%。该方法简单、快速、准确,适用于对大批量虾类样品中兽药残留的快速筛查,具有较高的实际应用价值。
