Ca3 Ti2 O7 杂化非本征铁电体的制备及其掺杂改性研究进展
2021-11-04陈大凯吴红迪符春林
陈大凯 ,蔡 苇,2 ,周 创 ,吴红迪,符春林,2
(1.重庆科技学院 冶金与材料工程学院,重庆 401331;2.纳微复合材料与器件重庆市重点实验室,重庆 401331)
人工智能、大数据等技术的飞速发展对信息存储材料的存储密度、速度、功耗等提出了更高的要求,基于铁电材料的非易失性存储器由于具有读写速度快(10 ns)、寿命长(1014次)和功耗低等优点引起了研究人员的广泛关注[1-4]。而同时具有铁电性和(反)铁磁性的多铁材料由于具有磁电耦合效应,可以实现多种逻辑状态,有望使存储密度显著提升,在多态存储器等方面具有重要的应用价值[5-11]。传统的单相多铁材料由于其极化和磁性产生机制,存在着磁电耦合弱[12]以及无法满足室温应用[13]等缺点。而非本征铁电性是由非极性畸变诱导产生,无需考虑传统铁电材料中极化和磁性电子构型的矛盾,更有利于获得具有更强磁电耦合的单相多铁材料[14-15]。近年来,具有Ruddlesden-Popper(R-P,An+1BnO3n+1,n=2)层状钙钛矿结构的等材料的杂化非本征铁电性(Hybrid Improper Ferroelectricity,HIF)逐渐引起关注,其铁电极化主要是由氧八面体的面内旋转(a0a0c+)和面外倾侧(a-a-c0)两种非极性畸变耦合引起的,这种新的极化来源为寻找新的单相多铁材料提供了重要途径。上述R-P 结构杂化非本征铁电体中,Ca3Ti2O7因在室温下具有较大的剩余极化强度(单晶在室温下的剩余极化强度约为8 μC/cm2)而备受关注,但其单晶和陶瓷的矫顽场强均较大。为进一步改善Ca3Ti2O7材料的铁电性,国内外学者在其制备方法及工艺优化、掺杂改性等方面开展了一系列的探索工作。为此,本文对近年来Ca3Ti2O7杂化非本征铁电体的制备及其掺杂改性的研究进展进行了综述,总结了Ca3Ti2O7材料的制备方法、工艺和阳离子取代对其性能影响的规律,并对未来研究的重点进行了展望。
1 Ca3Ti2O7的制备
Benedek 等[22]早在2011 年便采用第一性原理预测出Ca3Ti2O7材料具有杂化非本征铁电性,其自发极化强度可达20 μC/cm2,但是在相当长的一段时间里并未得到实验证实。随着国内外学者的不懈努力,Ca3Ti2O7单晶、陶瓷、薄膜被成功制备,其杂化非本征铁电性在实验上也逐步得到了证实。
1.1 Ca3Ti2O7单晶
2015 年,Oh 等[23]率先采用光学浮区法成功生长出Ca3Ti2O7单晶,在室温下采用双波法(Positive-Up-Negative-Down,PUND)测得其电滞回线(如图1(a)所示,剩余极化强度高达8 μC/cm2,矫顽场强为120 kV/cm),首次在实验上证实了Ca3Ti2O7的杂化非本征铁电性。迄今为止,Ca3Ti2O7单晶在室温下的剩余极化强度仍是R-P 层状结构杂化非本征铁电体中最高的。
1.2 Ca3Ti2O7陶瓷
相对于单晶而言,陶瓷不仅更容易制备,而且成本较低。Liu 等[24]采用固相反应法制备出Ca3Ti2O7陶瓷,在室温下测得了理想的电滞回线,但和单晶相比,陶瓷的剩余极化强度仅为0.6 μC/cm2(100 Hz),且矫顽场强较高(120 kV/cm),与单晶基本相当。Hu 等[25]采用相同的制备方法,对烧结工艺进行了改进:烧结温度升高了100 ℃,烧结时间缩短了36 h,增加了1400 ℃/3 h 氧气气氛下退火处理,获得了室温剩余极化强度为0.91 μC/cm2的Ca3Ti2O7陶瓷。作者所在课题组[26]采用1400 ℃烧结12 h 后再在1500 ℃下烧结12 h 的两次固相烧结法制备Ca3Ti2O7陶瓷,剩余极化强度达到1.319 μC/cm2(10 Hz),且矫顽场强较低(78.17 kV/cm)。Wu 等[27]采用固相反应法制备出的Ca3Ti2O7陶瓷剩余极化强度达1.6 μC/cm2(如图1(b)所示),该值为目前固相反应法制备的陶瓷的最高值,但其矫顽场强仍然较高(120 kV/cm)。综上,虽通过优化固相反应工艺使Ca3Ti2O7陶瓷的杂化非本征铁电性有一定程度提升,但其室温剩余极化强度仍然相对较低。与固相反应法相比,溶胶-凝胶法具有组分控制容易、均匀性好等优点。因此,为了进一步改善Ca3Ti2O7陶瓷的铁电性,作者所在课题组[28]率先以酒石酸为螯合剂,采用溶胶-凝胶法和固相烧结相结合,成功制备出单相Ca3Ti2O7陶瓷,采用PUND模式在10 Hz 和100 Hz 下分别测得其室温剩余极化强度高达4.32 μC/cm2和3.15 μC/cm2(如图1(c)所示),这是迄今为止文献报道的Ca3Ti2O7陶瓷的剩余极化强度最高值,这说明溶胶-凝胶法辅助固相烧结是获得优异杂化非本征铁电性Ca3Ti2O7陶瓷的一种有效途径。
1.3 Ca3Ti2O7薄膜
Li 等[29]采用脉冲激光沉积法首次在(110)SrTiO3衬底上制备出(010)取向的Ca3Ti2O7薄膜,发现:薄膜的铁电极化沿面内a轴方向,在2 K 下的剩余极化强度约为8 μC/cm2(与单晶相当),但其矫顽场强仅为5 kV/cm,远低于Ca3Ti2O7单晶和陶瓷(如图1(d)所示)。他们认为这种极低的矫顽场强是由薄膜中的非完美晶格所引起的。Shi 等[30]同样采用脉冲激光沉积法在(111)Pt 衬底上制备出Ca3Ti2O7薄膜,采用压电力显微镜证实了薄膜中电畴的翻转行为,并测得其压电系数吴曼[31]采用溶胶-凝胶匀胶法在(110) SrTiO3衬底上制备出具有b轴择优取向的Ca3Ti2O7薄膜,但在室温下并未测得明显的铁电性。虽然溶胶-凝胶匀胶法是制备Ca3Ti2O7薄膜的简便方法,但仍需进一步探索制备工艺与其微结构的关系,从而使其在室温下具有优异的铁电性。已有文献报道的Ca3Ti2O7材料的铁电性如表1 所示。
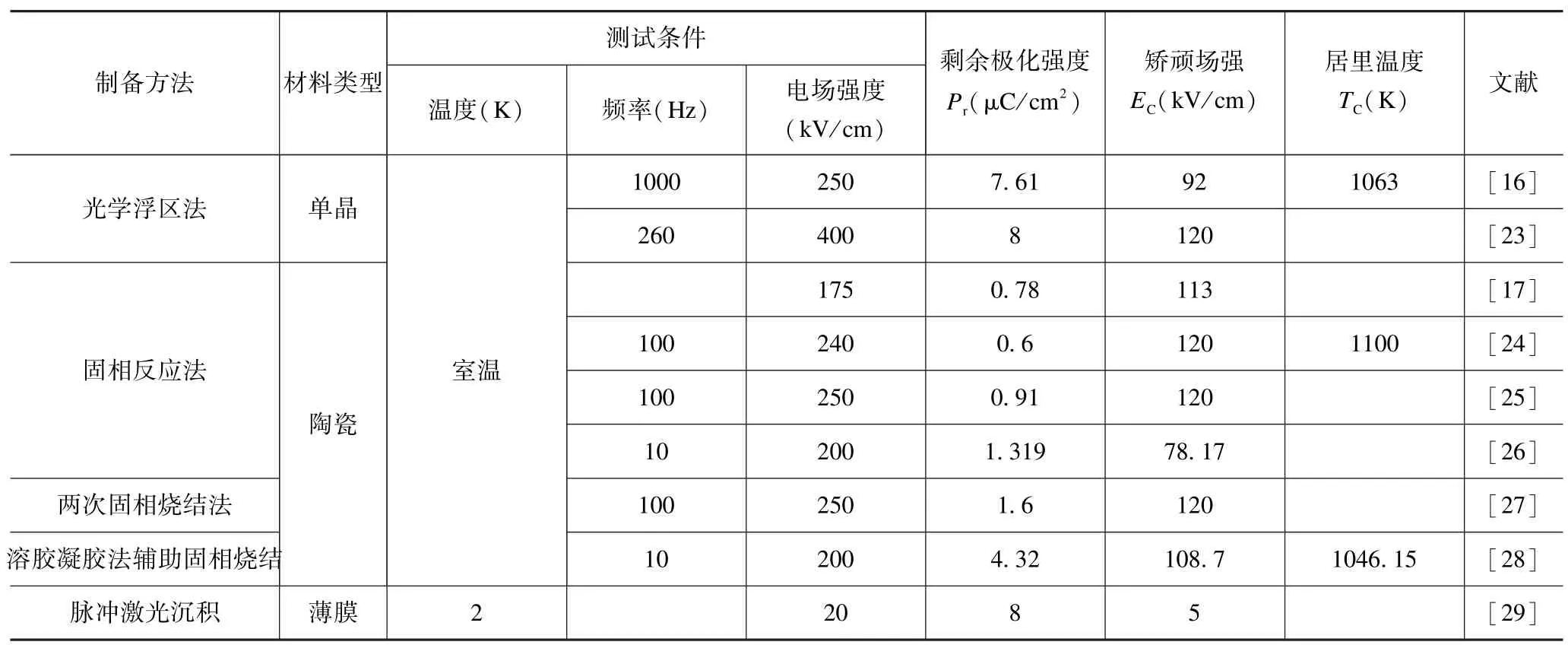
表1 Ca3Ti2O7材料的铁电性(PUND 模式)Tab.1 Ferroelectricity of Ca3Ti2O7 materials (PUND mode)
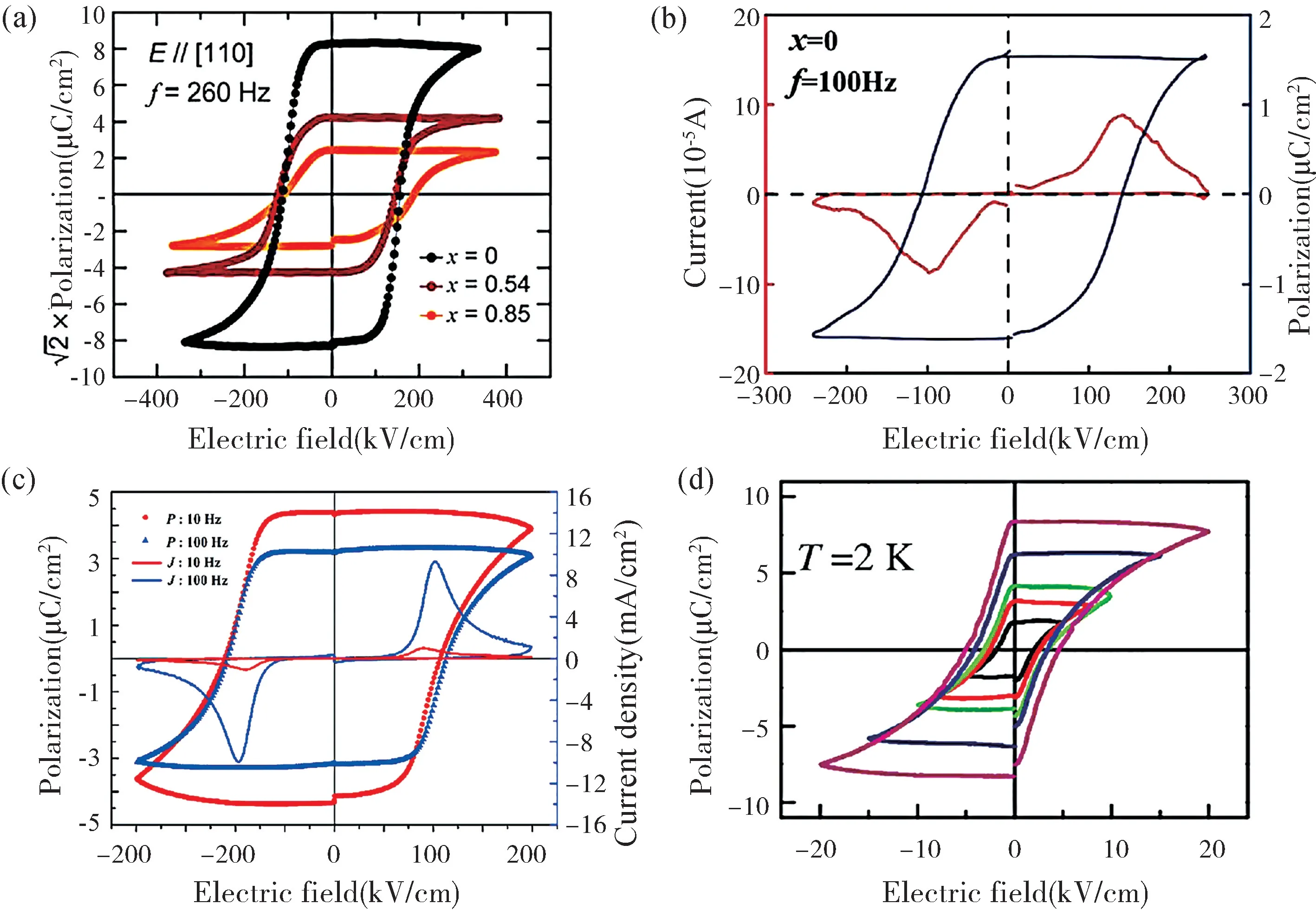
图1 Ca3Ti2O7基材料在PUND 模式下测得的电滞回线。(a) (Ca1-xSrx)3Ti2O7单晶[23];(b) 固相反应法制备的Ca3Ti2O7陶瓷[27];(c) 溶胶-凝胶法制备的Ca3Ti2O7陶瓷[28];(d) Ca3Ti2O7薄膜[29]Fig.1 Ferroelectric hysteresis loops of Ca3Ti2O7-based materials obtained by PUND mode.(a) (Ca1-xSrx)3Ti2O7 single crystal[23];(b) Ca3Ti2O7 ceramics fabricated by solid-state method[27];(c) Ca3Ti2O7 ceramics fabricated by sol-gel method[28];(d) Ca3Ti2O7 thin films[29]
2 掺杂对Ca3Ti2O7材料铁电性的影响
掺杂是提升材料性能的常用手段,由于具有R-P层状钙钛矿结构的Ca3Ti2O7材料的杂化非本征铁电性来自于A 位离子的反铁电畸变位移,而且其铁电极化是由TiO6氧八面体的面内旋转和面外倾侧耦合所导致[14-15,32]。因此,通过在Ca3Ti2O7的A 位和B 位引入金属阳离子有望实现对其杂化非本征铁电性的调控。Liu 等[33]采用第一性原理计算发现:Ca3Ti2O7材料钙钛矿层的容差因子对其杂化非本征铁电性具有重要影响,这进一步说明在A、B 位引入阳离子对铁电性具有重要的调控作用。目前,关于Ca3Ti2O7材料的掺杂改性研究主要有A 位掺杂、B 位掺杂和A/B 位共掺三种情况。
2.1 A 位掺杂
2013 年,Mulder 等[32]采用第一性原理研究了RP 结构的A3B2O7材料中A 位阳离子有序对铁电性的影响,发现:岩盐层中的A 位离子与钙钛矿层中的A’位离子之间的未抵消反铁电畸变位移造成了宏观的净极化,且可通过调控A 位离子的半径来改善铁电性。Oh 等[23]研究Ca3-xSrxTi2O7单晶时发现:随着Sr2+含量增加,其矫顽场强变化不大,但剩余极化强度却明显降低(如图1(a)所示)。Li 等[34]在Ca3-xSrxTi2O7陶瓷中发现了同样的现象,其电滞回线如图2(a)所示。他们认为由于TiO6氧八面体面内旋转和面外倾侧的幅度随Sr2+含量增加而减小,导致其剩余极化强度减小。Pomiro 等[35]制备出Ca3-xSrxTi2O7陶瓷,结合对称性以及第一性原理计算,从相转变揭示出Ca3-xSrxTi2O7陶瓷的物理特性与特定对称性、氧八面体旋转和倾侧的关系:Ca3-xSrxTi2O7在x=0 时表现出明显的从铁电A21am 到Acaa 的一级相变,当x=0.85 时则是铁电A21am 到P42/mnm 的连续二级相变;Sr2+引入后导致Ca3-xSrxTi2O7从一级相变到二级相变的改变,与TiO6八面体旋转和倾侧的动态运动有关,二级相变的产生伴随着氧八面体旋转幅度和倾侧幅度的降低,这很可能是Sr2+导致其剩余极化强度下降的原因。吴曼[31]研究A 位Sr2+对Ca3Ti2O7薄膜影响时发现:离子半径较大的Sr2+引入后降低了氧八面体的旋转和倾侧幅度,导致了极化的减弱。Li 等[36]采用第一性原理计算发现:将Mg2+引入到Ca3Ti2O7的A 位将显著增加剩余极化强度(从10.52 μC/cm2提升到18.48 μC/cm2),认为这是因为离子半径相对较小的Mg2+倾向于取代岩盐层的A 位离子,从而导致了更大的反铁电畸变位移。除了Sr2+、Mg2+等等价取代外,国内外学者对A 位异价取代如Na+[37-38]开展了研究,异价掺杂会引入点缺陷继而影响其性能,从缺陷化学反应角度看,Na+对A位Ca2+的部分取代会使氧空位浓度升高,一般会产生畴壁钉扎效应,使铁电畴不易翻转而引起矫顽场强增大。Jiang 等[37]制备出Ca3Ti2O7陶瓷和Ca2.99Na0.01Ti2O7陶瓷,测得其矫顽场强分别为60 kV/cm 和80 kV/cm。但Huang 等[38]制备出Ca3-xNaxTi2O7陶瓷,却发现:Na+的引入使矫顽场强由120 kV/cm 下降到50 kV/cm。虽然Na+对Ca3Ti2O7陶瓷矫顽场强的影响尚存在争议,但他们[37-38]均发现Na+的引入并未使其剩余极化强度得到提升。综上,虽然通过理论计算[36]发现了一些可提升Ca3Ti2O7材料剩余极化强度的A 位离子,但目前并未能从实验上得到证实,目前尚未找到可显著改善Ca3Ti2O7材料杂化非本征铁电性的A 位离子。
2.2 B 位掺杂
Liu 等[33]采用第一性原理研究发现:容差因子对Ca3Ti2O7铁电性能有重要影响,通过在B 位引入比Ti4+半径更大的金属阳离子(使其容差因子降低)可以实现铁电性的改善。早在他们提出这个影响关系之前,Liu 等[24]就已制备出了Ca3(Ti1-xMnx)2O7陶瓷,发现:当Mn4+含量为摩尔分数5%时,剩余极化强度变化不大,但随Mn4+含量增加,其剩余极化强度明显降低(如图2(b)所示),由此可见:在B 位引入离子半径更小的离子使容差因子增加,会导致其剩余极化强度的降低。吴曼[31]在Ca3Ti2O7薄膜的B 位引入Mn4+同样未能获得更好的铁电性。Zhang 等[39]分别制备出B 位单掺Al3+、共掺Al3+和Nb5+、共掺Al3+和Ta5+的Ca3Ti2O7陶瓷,发现在Ca3Ti2O7陶瓷的B 位进行异价阳离子取代可有效调控其铁电性:Ca3Ti1.8Al0.1Nb0.1O7陶瓷在10 Hz 和350 kV/cm 条件下测得的剩余极化强度Pr为2.13 μC/cm2,明显高于Ca3Ti2O7陶瓷,认为主要是三方面原因引起的:Nb np 轨道与O 2p 轨道的杂化增强了O2-和Nb5+的极化率,提高了总极化率;Nb—O 更长的共价键键长增强了NbO6八面体的体积畸变程度,进而增大了氧八面体的畸变程度;掺杂引起晶粒尺寸增大从而增强了铁电极化。而Ca3Ti1.8Al0.1Ta0.1O7和Ca3Ti1.9Al0.1O6.95陶瓷的剩余极化强度约为0.5 μC/cm2,前者主要是因为Ta5+比Nb5+多一个外层电子层,Ta5+的共价键重叠较大,极化率较低导致剩余极化强度减小,而后者的剩余极化强度减小则是由于Al3+的引入使其八面体倾斜幅度急剧下降所致。在Ca3Ti1.9Al0.1O6.95陶瓷中,离子半径更小的Al3+的引入(0.535 nm)引起容差因子增加,使得剩余极化强度有所下降,这与掺入Mn4+情况一致。但值得注意的是:单从容差因子的角度而言无法解释Ca3Ti1.8Al0.1Nb0.1O7和Ca3Ti1.8Al0.1Ta0.1O7陶瓷剩余极化强度的差异,Ta5+和Nb5+的离子半径相同(0.64 nm)使得两种材料的容差因子是一样的,但这两种材料相较于Ca3Ti2O7而言,剩余极化强度却是一增一减的,这说明其杂化非本征铁电性除受容差因子影响外,实际上还受其他因素如掺杂离子极化率等的影响。Wu 等[27]制备出Ca3Ti1.9Ru0.1O7陶瓷,发现:Ru4+的引入显著提升了剩余极化强度(4.4 μC/cm2,这是目前掺杂体系的最高值),且矫顽场强有一定程度降低(100 kV/cm)(如图2(c)所示),他们认为可能是因为离子半径更大的Ru4+(0.62 nm)引入后使容差因子减小,且使氧八面体倾斜角度增大,同时掺杂后引起晶粒尺寸增加所致。这与Liu 等[33]的理论预测的结果是吻合的。综上,通过在Ca3Ti2O7材料B 位引入离子半径和离子极化率更大的金属阳离子能够改善其铁电性。

图2 掺杂对Ca3Ti2O7基陶瓷铁电性的影响(PUND 模式)。Fig.2 Effects of doping on ferroelectricity of Ca3Ti2O7-based ceramics (PUND mode). (a)
2.3 A/B 位共掺
除了在Ca3Ti2O7的A 位或B 位引入阳离子外,国内外学者对其A/B 位共掺也开展了研究。Hu 等[25]在A 位引入La3+,在B 位引入Al3+,制备出Ca3-xLaxTi2-xAlxO7陶瓷,发现:其剩余极化强度随掺杂量增加逐渐降低(如图2(d)所示),剩余极化强度降低是由于掺杂导致氧八面体倾斜和旋转振幅降低所致。作者所在课题组[40]研究(Ca1-xSrx)3(Ti1-yMny)2O7陶瓷时发现:随Sr2+、Mn4+含量增加,剩余极化强度逐渐降低,这同样是因为Sr2+、Mn4+抑制了氧八面体旋转。
表2 给出了已有文献报道的Ca3Ti2O7基材料掺杂改性的实验结果。可以看出:仅A 位掺Na+使矫顽场强有所降低(尚有一定争议),B 位掺Ru4+使剩余极化强度提升的同时矫顽场强降低,B 位共掺Al3+、Nb5+使剩余极化强度有所提升。表3 归纳了Ca3Ti2O7基材料的剩余极化强度与氧八面体畸变的关系,可以看出:在剩余极化强度较高的陶瓷样品中,Ca3Ti2O7陶瓷(Pr=4.32 μC/cm2)的氧八面体旋转角θR最大,而Ca3Ti1.9Ru0.1O7陶瓷(Pr=4.4 μC/cm2)的氧八面体倾侧角θT最大,但是它们相应的氧八面体倾侧角和旋转角却并非最大。由此可见,氧八面体旋转角和倾侧角两者之间可能存在竞争关系,但即便如此,也仍然印证了Ca3Ti2O7基材料的剩余极化强度与氧八面体畸变程度之间的规律:剩余极化强度与氧八面体畸变程度呈正比。
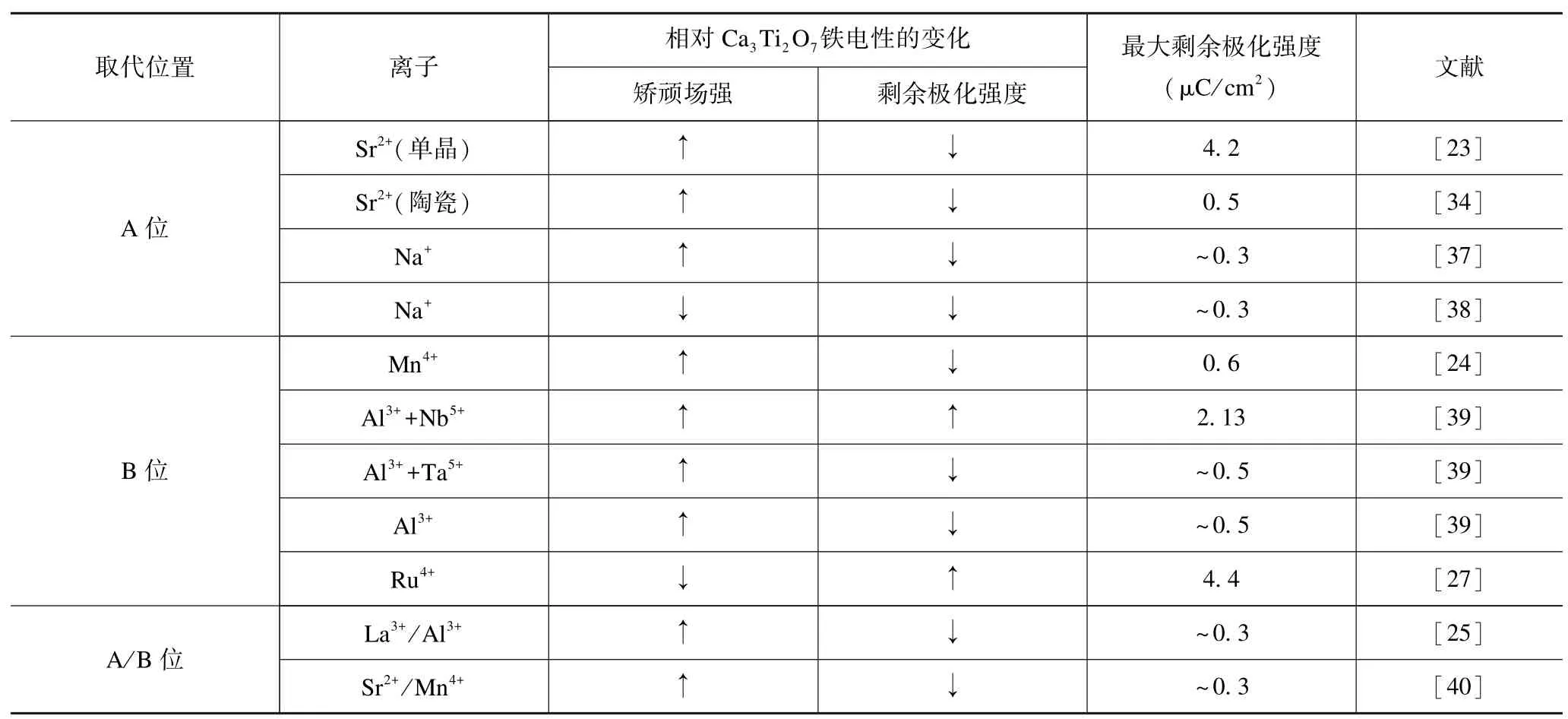
表2 掺杂改性对Ca3Ti2O7基材料铁电性的影响Tab.2 Influences of doping on ferroelectricity of Ca3Ti2O7-based materials
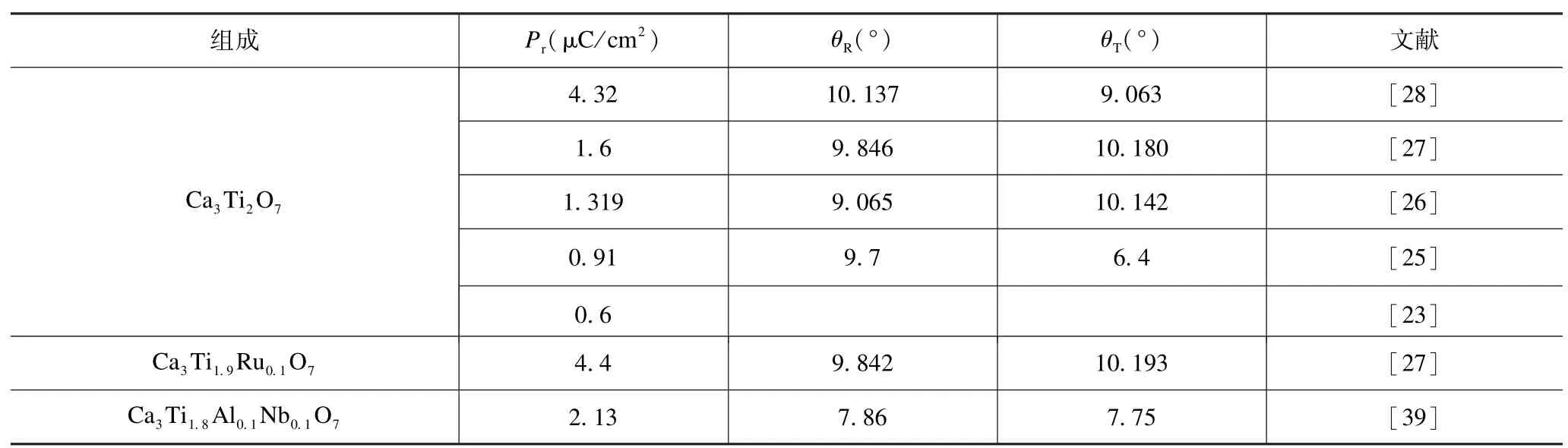
表3 Ca3Ti2O7基陶瓷氧八面体旋转/倾侧角度与剩余极化强度的关系Tab.3 Relationships between oxygen octahedral rotation/tilting angles and the remnant polarization of Ca3Ti2O7-based ceramics
3 结束语
R-P 层状钙钛矿结构的Ca3Ti2O7基材料因其独特的铁电极化产生方式,有望成为新的室温单相多铁材料,对其制备方法及工艺进行优化、掺杂改性研究具有重要的意义。本文对Ca3Ti2O7基材料的制备和掺杂改性进行了概述,归纳总结了近年来Ca3Ti2O7材料制备方法及工艺优化、掺杂改性的相关研究进展,并对其改性机理进行了详细阐述。国内外已有结果表明:采用化学法如溶胶-凝胶法制备Ca3Ti2O7陶瓷可获得明显优于固相反应法的铁电性;而B 位引入Ru4+、B位同时引入Al3+和Nb5+可使其剩余极化强度得到提升。但Ca3Ti2O7材料在室温下的铁电性仍需进一步提升,重点开展以下方面工作。
(1)探索并优化溶胶-凝胶法、水热法、共沉淀法、熔盐法等制备Ca3Ti2O7基材料的工艺,进一步提升其杂化非本征铁电性。
(2)氧八面体的畸变程度是影响Ca3Ti2O7材料杂化非本征铁电性重要的因素,但引入金属阳离子后对氧八面体旋转和倾侧的影响有时是存在差异的。因此,需进一步系统开展Ca3Ti2O7材料掺杂改性研究,探索构筑A 位阳离子有序与降低容差因子结合的方式调控铁电性的可能。
(3)结合第一性原理计算,探究除容差因子、离子极化率、氧八面体畸变程度等以外的Ca3Ti2O7基材料铁电性的其他影响因素,从而阐明其杂化非本征铁电性调控机制。
