紫外分光光度法快速检测百草枯中毒
2021-11-02陈姿如杜书明
陈姿如,杜书明
百草枯(paraquat, PQ),是一种快速灭生性除草剂,可经由皮肤、黏膜、呼吸道、消化道等进入人体,以肺部分布最多,损伤最大,肺纤维化伴呼吸衰竭是患者中毒后期死亡的主要原因,且因其在临床上没有特效解毒的手段,病死率高达80%以上[1,2]。我国2016年7月禁止生产和使用百草枯,但临床上百草枯中毒案例依旧存在。中毒患者体内百草枯浓度与病死率有着密切的关系,测定患者血浆中百草枯浓度不仅有助于准确诊断,并对制定合理的治疗方案有重要意义。本实验旨在探讨使用合适的样品前处理方法,建立灵敏、快速检测百草枯中毒的紫外分光光度法,为基层医院快速诊断百草枯中毒提供帮助。
1 材料与方法
1.1 仪器与试剂 采用UV-2600紫外/可见分光光度计(上海天美科学仪器有限公司);7820气相色谱仪(山东鲁南瑞红化工仪器有限公司),氮磷检测器(NPD),DB-5毛细色谱柱(30 m×0.23 mm×0.25 μm),LC-LX-H165A高速离心机。试剂:保险粉(连二亚硫酸钠),碳酸氢钠,饱和氢氧化钠溶液(现配),20%高氯酸甲醇溶液,20%硼氢化钠溶液,环己烷化学试剂均为分析纯,百草枯对照品购自上海甄准生物科技有限公司。
1.2 试验方法 抽取患者静脉血3 ml(同时做健康人对照),3000 r/min高速离心后,取0.5 ml血浆于带盖的5 ml塑料试管中,加新鲜配制的20%高氯酸甲醇溶液0.2 ml 充分振荡30 s混匀,以 12 000 r/min高速离心10 min。取上清液0.5 ml,加入碳酸氢钠0.2 mg碱化和连二亚硫酸钠0.5 mg显色,再加饱和氢氧化钠溶液0.1 ml增色,pH>14,采用50 μl微量比色池,设置200~700 nm波长自动扫描,以蒸馏水做基线测量,扫描待测液,百草枯吸收峰出现在紫外区(395±2)nm和可见光区(605±2)nm,空白液扫描不显色,无吸收峰,记录最大吸收峰(605±2)nm吸光度,查标准曲线或回归方程求出血浆中百草枯含量。另取0.5 ml血浆加入20%高氯酸甲醇溶液高速离心取上清液加强和氢氧化钠液0.1 ml,再加入20%硼氢化钠溶液0.5 ml,于室温反应30 min,加入环己烷0.5 ml,振荡30 s,高速离心,取上清液作为检材样品衍生液,供气相色谱分析或气相色谱质谱法分析。
1.3 监测指标及标准 检验血液中的百草枯,按我国公安部发布的标准(GA/T1629-2019)执行。检测、工作曲线、检出限及回收率和精密度试验结果,并对百草枯紫外测定方法的特异性和稳定性进行分析。
1.4 检测波长的选择 配制0、5.0、10.0、15、20.0 μg/ml的百草枯水溶液,以蒸馏水做空白,取5个带盖的5.0ml塑料试管加不同浓度百草枯水溶液0.5 ml,空白管加蒸馏水0.5 ml,然后按照1.2血样处理及试验方法操作,百草枯最大吸收峰为(605±1)nm,以最大吸收峰定性定量。同时配置0.0、5.0、10.0、20.0μg/ml的百草枯血浆标本,按照1.2血样处理及试验方法操作,结果百草枯扫描可见光最大吸收光谱在(605±1)nm,血浆中的百草枯同样在(605±2)nm处有最大吸收峰。
1.5 工作曲线和检出限 精确称取百草枯对照品100 mg置于100 ml定量瓶中,加蒸馏水配成1 mg/ml的百草枯标准贮存液,贮存液采用健康人血浆配制为0.05、0.10、0.50、1.0、5.0、10.0、20.0、40 μg/ml标准工作液,按照1.2血样处理及试验方法操作。根据吸光度值(y)和对应的浓度(x)绘制标准曲线,百草枯浓度在0.05~40.0 μg/ml范围内线性关系良好,回归方程(y)=0.0896+0.0102,相关系数(r)为0.9992,检测下限为0.05 μg/ml。
1.6 回收率和精密度试验 采用空白血样加标的方式进行回收试验,分别添加0.5、1.0、5.0、10.0 μg/ml共四个水平,每个水平单独测定5次,回收率为85.0%~94.0%,变异系数均小于4.8%,见表1。
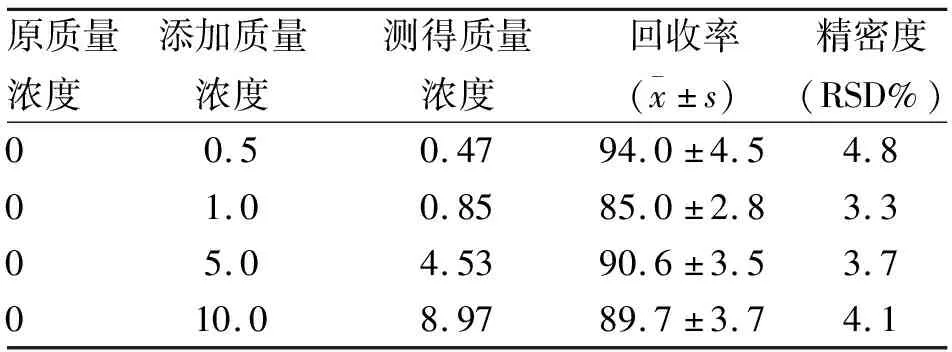
表1 百草枯回收率和精密度试验 (n=5;μg/ml)
1.7 百草枯测定方法的特异性和稳定性 选取百草枯中毒患者与健康人血浆各20人份,为中毒组与对照组,均加入20%高氯酸甲醇溶液高速离心,分离血浆蛋白,操作按照1.2血样处理及试验方法进行。中毒组的血浆中百草枯最大吸收峰均为(605±1)nm,对照组不显色,扫描无吸收峰,表明方法的特异性良好。上清液加入连二亚硫酸钠、碳酸氢钠后显蓝色,但蓝色会很快褪色,当加入饱和氢氧化钠液后,会明显增加蓝色深度,提高了试验方法的灵敏度。稳定性试验:每隔2 min扫描一次吸光度,观察吸光度的变化,10 min内无褪色,10 min后开始逐渐褪色,表明该试验在显色10 min内显色稳定,所以应在10 min内完成比色,即可提高试验结果的精密度和准确度。
1.8 监测指标及标准结果对比 为了证实该方法的准确性、可靠性,配置0.0、1.0、5.0、10.0 μg/ml的百草枯血浆标本,按1.2样品处理及试验方法操作。同时按照GA/T1629-2019监测指标及标准配制试剂,对同一样品进行衍生化气相色谱法分析和百草枯显色紫外分光光度法分析,结果见表2。

表2 不同检测方法测定血液百草枯的结果
2 讨 论
碱性的连二亚硫酸钠可将百草枯还原成蓝色的自由基,临床上利用百草枯这一理化性质,已应用于尿液中的百草枯测定[3-6]。由于血液单位体积内百草枯含量明显低于尿液[7],且送检血液量也较少,且误服百草枯后送检样本多为血液,血液中的百草枯测定往往需要气相色谱法,液质联用法、高效液相色谱法等贵重仪器检测,基层医院实验室很难做到,采用常规百草枯显色测定,准确性较差。所以如何准确、快速检测血液中的百草枯含量,对临床救治非常重要。本研究对传统血液百草枯测定方法进行了改进和探讨,在血液离心后取血浆0.5 ml加20%高氯酸甲醇溶液后再次高速离心,上清液用碳酸氢钠碱化和连二亚硫酸钠显色后,再加饱和氢氧化钠溶液0.1 ml增色,pH>14,使血液中的百草枯测定灵敏度明显提高,显色稳定性增强,结果显示,饱和氢氧化钠溶液对百草枯测定方法的稳定性、灵敏度至关重要,只要准确把握显色时间,在10 min内完成比色,即可保证试验方法的准确性。
本研究还以蒸馏水做基线测量,在200~700 nm波长自动扫描百草枯,可以排除血浆基质对基线测量的干扰,防止出现假阳性或假阴性的结果。结果发现,百草枯吸收峰出现在紫外区(395±2)nm和可见光区(605±2)nm。选择(605±2)nm为定性定量主峰,按本方法操作一旦在(605±2)nm处出现最大吸收峰,就表明血液中百草枯成分的存在。本研究通过百草枯回收率和精密度试验显示:样品加标回收率为85.0%~94.0%,方法变异系数均小于4.9%,表明能够满足临床中毒诊断的需要。百草枯浓度在0.05~40.0 μg/ml范围内线性关系良好,定量检测下限为0.05 μg/ml。
气相色谱法、气质联用法、高效液相色谱法等采用的仪器价格昂贵,样品前处理繁琐用时长,不适合突发事件的检测,致使基层医院乃至三甲医院开展血液百草枯测定受限[4-6]。2018—2020年我院毒检科共检测265例百草枯中毒患者,误服中毒92.1%(244/265),意外中毒7.9%(21/265)。并抽取其中的100例进行两种方法对比试验,结果证明采用紫外分光光度法与气相色谱法测定百草枯浓度检测结果相同,数据准确可靠,无1例漏诊误诊。证明紫外分光光度法准确性高,且避免了气相色谱衍生化中繁琐的操作过程[8],操作快速、简便, 值得临床推广应用。
