河北省致犊牛腹泻主要病原多重PCR检测方法的建立
2021-10-30高亚桃刘立元郭姚蕊刘胜丽马亚宾蒋桂娥刘志勇秦建华赵月兰河北农业大学动物医学院河北保定0700河北省畜牧良种工作站河北石家庄05006河北省唐山市动物疫病防控中心河北唐山06400
高亚桃,李 妍,刘立元,王 猛,郭姚蕊,刘胜丽,马亚宾,蒋桂娥,刘志勇,秦建华*,赵月兰* (.河北农业大学 动物医学院,河北 保定 0700;.河北省畜牧良种工作站,河北 石家庄 05006;.河北省唐山市动物疫病防控中心,河北 唐山06400)
近年来,随着养殖业的不断发展,犊牛腹泻引起的传染病在我国广泛流行。多种病原可引起犊牛腹泻,导致犊牛发生细菌性、寄生虫性和病毒性腹泻等[1]。由病毒引起的犊牛腹泻较为严重,引起犊牛消化道疾病的主要病毒有牛病毒性腹泻病毒(bovine viral diarrhea virus,BVDV)、牛轮状病毒(bovine rotavirus,BRV)、牛传染性鼻气管炎病毒(infectious bovine rhinotracheitis virus,IBRV)等[2-3]。 BVDV是引起牛病毒性腹泻/黏膜病的主要病原,各个年龄段的牛均易感,尤其是3~5周龄犊牛最易感,该病毒主要感染黄牛和奶牛,病情严重时可导致患病牛死亡[4-5]。BRV是引起新生犊牛腹泻的重要病原,又称为“犊牛腹泻病毒”[6-7],15日龄内犊牛对该病毒最易感,感染BRV的犊牛主要表现为精神沉郁、排水样稀粪,多可引起继发感染,发病急、死亡率高[8]。IBRV是引起牛肠炎型腹泻的重要病原之一,可引起牛的急性、热性、接触性传染病[9],感染该病毒的牛主要表现为呼吸道症状,患病牛有时出现带血性腹泻,新生犊牛可表现为全身性疾病。BVDV、BRV和IBRV这3种病毒均可导致牛腹泻、呼吸障碍、流产及犊牛免疫力低下等[10-11],是引起犊牛腹泻的重要病原,严重危害犊牛健康,给养牛业造成巨大的经济损失。目前对于这3种病毒的检测主要依赖于病毒的分离鉴定、ELISA检测方法、PCR检测方法以及荧光定量PCR检测方法等[12],其中PCR检测方法较简单、快速、准确性高、特异性强。目前,对于这3种病毒同时进行检测的多重PCR检测方法还未见报道。为了能快速、同时检测出BVDV、BRV和IBRV 3种病毒,本研究建立了一种多重PCR检测方法,为犊牛腹泻病的诊断和流行病学的调查提供一种有效的检测手段。
1 材料与方法
1.1 毒株和病料BVDV、BRV、IBRV、牛冠状病毒(bovine coronavirus,BCoV)、牛细小病毒(bovine parvovirus,BPV)毒株和牛隐孢子虫虫种,均由河北农业大学动物医学院预防兽医学实验室分离、鉴定和保存。临床检测样品为从河北省7个地区的15个养殖场采集的50份腹泻犊牛粪便。
1.2 主要试剂DNA/RNA提取试剂盒、琼脂糖凝胶DNA纯化回收试剂盒和质粒小提试剂盒购自上海汇易生物科技有限公司;反转录试剂盒购自南京诺唯赞生物科技股份有限公司;DL2000 DNA Marker购自天地人和生物科技有限公司;DH5α感受态细胞和pUC57载体购自吉林省库美生物科技有限公司。
1.3 主要仪器PCR扩增仪和NanoDrop 2000 超微量分光光度计购自赛默飞世尔科技公司;电泳仪购自北京六一仪器厂;化学发光成像系统购自美国ProteinSimple公司;离心机购自上海昆士兰生物科技发展有限公司。
1.4 引物设计与合成根据GenBank中登录的BVDV的5′-UTR序列、BRV的VP6基因和IBRV的gB基因,利用Primer Premier 5.0软件设计3对特异性引物,由吉林省库美生物科技有限公司合成,引物序列及相关信息见表1。

表1 引物序列及相关信息
1.5 病毒DNA/RNA的提取将保存在-80℃的病毒取出,反复冻融3次后,利用上海汇易生物科技有限公司的OMEGA E.Z.N.A Viral DNA/RNA Kit提取病毒DNA/RNA,将提取的IBRV的DNA保存在-20℃,BVDV和BRV的RNA保存在-80℃备用。
1.6 反转录cDNA的合成将1.5中提取的BVDV和BRV的RNA,利用南京诺唯赞生物科技股份有限公司的HiScript®Ⅲ 1st Strand cDNA Synthesis Kit进行反转录,反转录产物置于-20℃保存备用。
1.7 PCR扩增以提取的BVDV和BRV的cDNA以及IBRV的DNA为模板,分别用相应引物进行单一PCR扩增,用无菌去离子水设立阴性对照。反应体系:2×Phanta Max Master Mix 12.5 μL,ddH2O 9.5 μL,上、下游引物各1 μL,DNA/cDNA模板1 μL,总反应体系25 μL。反应条件:94℃ 5 min;94℃ 30 s,57℃ 15 s,72℃ 15 s,共35个循环;72℃ 7 min。扩增反应结束后,取5 μL PCR产物通过2%琼脂糖凝胶电泳进行检测,用琼脂糖凝胶纯化回收试剂盒回收特异性条带,回收产物送吉林省库美生物科技有限公司进行测序。
1.8 多重PCR退火温度的优化以1.7中PCR反应的退火温度为基准,每上下递减1℃作为退火温度,同时用无菌去离子水设立阴性对照。反应体系:2×Phanta Max Master Mix 12.5 μL,3种病毒的DNA/cDNA模板各1 μL,3对引物上下游各1 μL,ddH2O 3.5 μL。反应程序:94℃ 5 min;94℃ 30 s,54,55,56,57和58℃ 15 s,72℃ 15 s,共35个循环;72℃ 7 min。扩增反应结束后,取5 μL反应产物进行琼脂糖凝胶电泳,筛选出最佳退火温度。
1.9 多重PCR特异性试验利用优化的PCR反应体系,以牛隐孢子虫、BPV和IBRV的DNA,BVDV、BRV和BCoV的cDNA为模板,用表1中的3对引物,按1.8筛选出的最佳退火温度进行扩增。PCR结束后取5 μL产物通过2%琼脂糖凝胶电泳检测,利用琼脂糖凝胶纯化回收试剂盒回收特异性扩增条带。
1.10 多重PCR灵敏性试验将1.9中得到的3种病毒特异性片段的胶回收产物送吉林省库美生物科技有限公司进行测序鉴定,将序列正确的产物分别克隆至pUC57载体,获得阳性对照质粒pUC57-UTR、pUC57-VP6和pUC57-gB。用NanoDrop 2000超微量分光分度计测定质粒浓度,根据公式计算质粒拷贝数。将3种病毒的阳性对照质粒10倍倍比稀释至1×101copies/μL,以不同质粒拷贝数的阳性对照质粒为模板,按照优化好的反应条件和反应体系进行PCR扩增。PCR反应结束后,取5 μL PCR反应产物于2%琼脂糖凝胶中进行电泳,检测所建立的多重PCR方法的灵敏性。
1.11 多重PCR重复性试验利用已建立的多重PCR检测方法,以构建的阳性对照质粒为模板进行PCR扩增,以相同反应条件和反应体系,在不同时间重复扩增3次,检测该方法的重复性。
1.12 临床样品的检测利用建立的多重PCR检测方法,对从河北省部分地区15个养牛场采集的50份粪便样品进行检测。同时与单一PCR检测方法进行比较,并计算两种方法的符合率,符合率=(真阳性数+真阴性数)/被检总数。
2 结果
2.1 3种病毒PCR扩增结果以BVDV和BRV的cDNA和IBRV的DNA为模板,分别用相应的引物进行PCR扩增。扩增结果如图1所示,扩增出的目的片段分别为189,144,109 bp,与预期大小相符。PCR产物测序后与GenBank中登录的序列进行比对,结果显示BVDV扩增产物序列与BVDV(HB-BDZ)5′-UTR部分序列的同源性为96.8%~97.9%,其中与MN513409的同源性最高,为97.9%(图2);BRV扩增产物序列与BRV(HB-DZZ) VP6部分基因的同源性为96.5%~100.0%,其中与MN047454的同源性最高,为100%(图3);IBRV扩增产物序列与IBRV(HB-XTZ)gB部分基因同源性为98.2%~100.0%,其中与MK654723、MF-287966、MG497777和KU992439的同源性最高,为100.0%(图4)。

M.DL2000 DNA Marker;1.BVDV 5′-UTR序列;2.BRV VP6基因;3.IBRV gB基因;4.阴性对照图1 3种病毒PCR扩增结果
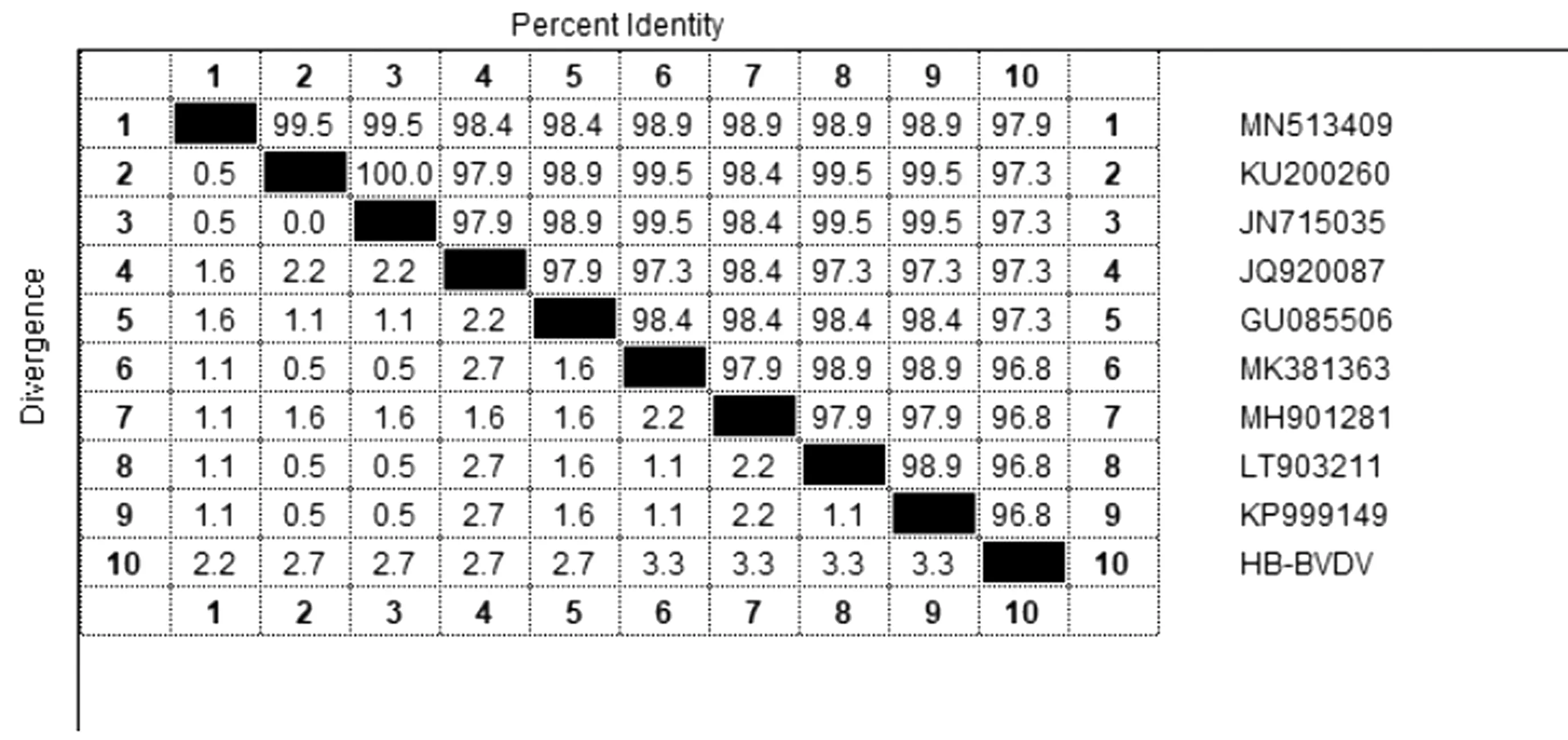
图2 BVDV 5′-UTR序列同源性比较结果
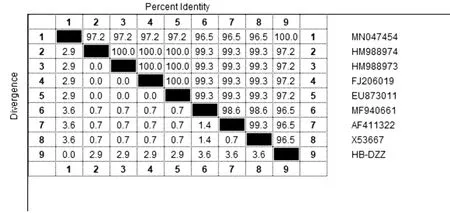
图3 BRV VP6基因同源性比较结果

图4 IBRV gB基因同源性比较结果
2.2 多重PCR退火温度优化结果分别以54,55,56,57,58℃为退火温度,对多重PCR退火温度进行优化,结果如图5所示,当退火温度为56℃时,3种病毒的扩增条带最亮,因此,将56℃ 15 s作为多重PCR检测方法的最佳退火温度和时间。最终确定多重PCR检测方法的最佳反应条件:3种病毒的DNA/cDNA模板各1 μL,3对引物上下游各1 μL,2×Phanta Max Master Mix反应液12.5 μL,ddH2O 3.5 μL,总体系25 μL。最佳反应程序:94℃ 5 min;94℃ 30 s,56℃ 15 s,72℃ 15 s,共35个循环;72℃ 延伸7 min。
2.3 多重PCR特异性试验结果分别以3种病毒的DNA/cDNA混合物、BVDV、BRV和BCoV的cDNA及IBRV、BPV、牛隐孢子虫的DNA和水为模板,用3对引物进行扩增,检测已建立的多重PCR反应体系中引物的特异性。结果如图6所示,以3种病毒DNA/cDNA混合物、BVDV和BRV的cDNA以及IBRV的DNA为模板,均可扩增出相应目的片段,而以BCoV的cDNA和BPV、牛隐孢子虫的DNA为模板均未扩增出条带,表明该检测方法具有良好的特异性。

M.DL2000 DNA Marker;1.3种病毒DNA/cDNA混合物;2.BVDV 5′-UTR序列;3.BRV VP6基因;4.IBRV gB基因;5.BCoV;6.BPV;7.牛隐孢子虫;8.阴性对照图6 多重PCR特异性试验结果
2.4 多重PCR灵敏性试验结果利用NanoDrop 2000超微量分光分度计测定3种阳性对照质粒浓度,根据公式计算确定pUC57-UTR的拷贝数为1.44×1011copies/μL,pUC57-VP6的拷贝数为1.38×1011copies/μL,pUC57-gB的拷贝数为1.2×1011copies/μL。将3种阳性对照质粒按照10倍倍比进行稀释,以不同质粒拷贝数梯度的阳性对照质粒为模板,按照已建立好的多重PCR检测方法进行扩增,检测该方法的灵敏性。结果如图7所示,该方法的检出限分别为BVDV 1.44×105copies/μL,BRV 1.38×105copies/μL和IBRV 1.2×105copies/uL,表明该方法具有良好的灵敏性。

M.DL2000 DNA Marker;1~10.模板量为1×10-2~1×10-11;11.阴性对照图7 多重PCR灵敏性试验结果
2.5 多重PCR重复性试验结果利用已建立的多重PCR检测方法对同一模板进行扩增,结果如图8所示,3次不同时间的重复试验结果一致,表明该方法具有良好的重复性。

A~C.3次不同时间的重复试验。M.DL2000 DNA Marker;1. 3种病毒DNA/cDNA混合物;2.BVDV 5′-UTR序列;3.BRV VP6基因;4.IBRV gB基因;5.阴性对照图8 多重PCR重复性试验结果
2.6 临床样品的检测结果利用常规PCR检测方法和本试验建立的多重PCR检测方法分别对50份临床样品进行检测。结果显示,单一PCR检测方法对BVDV、BRV和IBRV的检出阳性率分别为41.4%(27/50),40%(20/50),34%(17/50);多重PCR方法对BVDV、BRV和IBRV的检出阳性率分别为64%(32/50),56%(28/50),46%(23/50)。其中BVDV和BRV混合感染的检出率为 18%(9/50),BVDV和IBRV混合感染的检出率为14%(7/50),BRV和IBRV混合感染的检出率为22%(11/50),BVDV、BRV和IBRV 3种病毒混合感染的检出率为4%(2/50)。2种方法的总符合率分别为BVDV 90%,BRV 84%和IBRV 88%。结果表明,本研究所建立的多重PCR检测方法能快速检测出BVDV、BRV和IBRV 3种病原。
3 讨论
牛病毒性腹泻主要是由多种病毒引起的一种接触性传染性疾病,其中BVDV和BRV是引起犊牛腹泻的主要病原。IBRV临床上也可引起肠炎,犊牛有时甚至出现血便,严重危害犊牛健康[13-14]。临床上,这3种病毒常呈混合感染,临床症状难以区分,患病牛可终身带毒,且可引起其他病毒或细菌的继发感染,导致患病牛生产性能下降或死亡,严重危害牛群健康,给养牛业造成巨大的经济损失[15]。目前,对于这3种病原引起的犊牛腹泻,尚没有特效药物,因此,尽快检出病原,淘汰、隔离患病牛或带毒牛是控制该病传播的有效手段[16]。目前对于这3种病毒的检测还主要依赖于病毒分离鉴定、PCR技术和ELISA等方法,PCR检测技术由于具有操作简便、特异性高和灵敏性强的特点,在临床检测时常常被采用[17-22]。
本研究根据3种病毒高度保守的基因序列如BVDV的5′-UTR序列、BRV的VP6基因和IBRV的gB基因,经过对反应条件和退火温度的优化,建立了一种多重PCR检测方法。该方法对BCoV、BPV、牛隐孢子虫无交叉反应,具有良好的特异性;对BVDV最低的检测拷贝数是1.44×105copies/μL,对BRV最低的检测拷贝数是1.38×105copies/μL,对IBRV最低的检测拷贝数是1.2×105copies/μL,具有良好的灵敏性。利用已建立的多重PCR检测方法和单一PCR检测方法对从河北省15个地区采集的50份临床样品进行检测,发现两种检测方法的总符合率BVDV为90%,BRV为84%,IBRV为88%,且结果表明在秋冬季节,临床上犊牛以感染BVDV引起腹泻的概率最高。本研究建立的多重PCR检测方法特异性好、灵敏度高,可以应用到临床上对BVDV、BRV和IBRV的检测,为检测引起犊牛腹泻的重要病原提供一种有效的方法。
