扶正治癃颗粒提取、成型工艺的优化
2021-10-26马转霞孙岚萍顾志荣马天翔许爱霞
马转霞, 孙岚萍, 顾志荣, 马天翔, 吕 鑫, 许爱霞, 葛 斌
(1.甘肃中医药大学药学院,甘肃 兰州 730000;2.甘肃省人民医院药剂科,甘肃 兰州 730000)
扶正治癃方由黄芪、熟地、肉苁蓉3味中药组成,是甘肃省人民医院的临床经验方,具有补气温肾、补血养阴、补肾润燥等功效,常用于治疗前列腺增生等疾病。方中君以黄芪补气升阳、益气温肾、调节气血;臣以熟地填精益髓、补血养阴,肉苁蓉补肾、益精、润燥。全方以补肾为主,气血阴阳统调,标本兼治。研究发现,黄芪利尿机制为竞争性抑制Na+-K+-ATP酶活力,且黄芪皂苷可促进膀胱逼尿肌的收缩,缓解尿道内口括约肌紧张度,减轻前列腺增生排尿无力、排尿困难的症状[1]。熟地、肉苁蓉均含有黄酮、多糖、生物碱等多种化学成分,具有抑菌、抗炎、提高机体免疫的生物活性,可保护前列腺免遭细菌和其他病原微生物侵袭,解除尿路炎性梗阻,改善局部症状。
中医常将前列腺增生归为“癃闭”“淋证”等范畴,其发病基础为年老体衰、肾气亏虚,基本病理因素为瘀血、痰浊、湿热,常见发病条件为劳力过度、情志刺激、外感六淫、饮食不节等[2]。中药及复方制剂治疗该类疾病具有疗效确切、整体调节、毒副作用小、适合长期服用等诸多明显优势[3]。故拟将原汤剂制备成便于服用、携带与贮存的颗粒剂,以解决传统汤剂煎煮费时、繁琐,且药液味苦量大等问题。本研究采用正交试验、均匀设计优化提取及成型工艺,为扶正治癃颗粒的开发研究提供技术资料。
1 材料
1.1 仪器 LC-16型高效液相色谱仪(日本岛津公司);UV8100A型紫外-可见分光光度计(北京莱伯泰科仪器有限公司);BSA4202S-CW型电子天平(百万分之一,德国赛多利斯公司);HH-6型数显恒温水浴锅(西安超杰仪器有限公司);DHG-9140A型电热鼓风干燥箱(上海一恒科学仪器有限公司);DD-5M型低速大容量离心机(湖南湘仪离心机仪器有限公司);YC-1000型实验室喷雾制粒包衣机(上海雅程仪器设备有限公司)。
1.2 试剂与药物 黄芪(批号180606)、熟地(批号180602)、肉苁蓉(批号180201)均购于甘肃冠兰中药饮片有限公司,经甘肃中医药大学李硕副教授鉴定为正品,符合2015年版《中国药典》一部相关规定。5-羟甲基糠醛(批号CHB171016)、松果菊苷(批号CHB170302)、毛蕊异黄酮葡萄糖苷(批号CHB171102)、毛蕊花糖苷(批号CHB171103)、D-无水葡萄糖(批号CHB180115)、芦丁(批号CHB170303)对照品均购于成都克洛玛生物科技有限公司。预胶化淀粉、麦芽糊精、甘露醇、微晶纤维素均为药用规格。甲醇、甲酸为色谱纯(天津大茂化学试剂厂);其他试剂均为分析纯(天津大茂化学试剂厂);水为纯化水。
2 方法与结果
2.1 提取工艺优化
2.1.1 HPLC法测定4种成分含量
2.1.1.1 对照品溶液制备 精密称取5-羟甲基糠醛、松果菊苷、毛蕊异黄酮葡萄糖苷、毛蕊花糖苷对照品适量,置于25 mL量瓶中,甲醇超声溶解,定容至25 mL,即得(四者质量浓度分别为0.10、4.40、0.19、0.53 mg/mL)。
2.1.1.2 供试品溶液制备 取水煎液10 mL,置于100 mL具塞三角瓶中,精密加入15 mL甲醇,密塞,称定质量,超声(功率500 W、频率40 kHz、温度20 ℃)处理60 min,提取液冷却至室温,甲醇补足减失的质量,摇匀,滤过,取上清液,溶剂回收至干,残渣加纯化水溶解,转移至 25 mL量瓶中,纯化水洗涤并定容至刻度,即得。
2.1.1.3 色谱条件 WondaSil C18-WR色谱柱(4.6 mm×150 mm,5 μm);流动相甲醇(A)-水(含0.1%甲酸)(B),梯度洗脱(0~30 min,8%~62%A;30~32 min,62%A);体积流量1.0 mL/min;柱温40 ℃;检测波长290 nm;进样量10 μL。色谱图见图1。

1.5-羟甲基糠醛 2.松果菊苷 3.毛蕊异黄酮葡萄糖苷 4. 毛蕊花糖苷
2.1.1.4 线性关系考察 精密吸取“2.1.1.1”项下对照品溶液0.2、0.5、1.0、1.5、2.0、2.5、3.0 mL,置于10 mL量瓶中,甲醇定容至刻度,各取10 μL,在“2.1.1.3”项色谱条件下进样测定。以对照品质量浓度(mg/mL)为横坐标(X),峰面积为纵坐标(Y)进行回归,结果见表1,可知各成分在各自范围内线性关系良好。

表1 各成分线性关系
2.1.1.5 方法学考察 选择正交1号水煎液,按照2015年版《中国药典》四部指导原则进行精密度、稳定性、重复性、加样回收率试验,测得RSD均小于1.88%,各成分平均加样回收率在99.39%~102.26%之间,表明仪器精密度、方法重复性良好,水煎液在室温下24 h内稳定。
2.1.2 总多糖含量测定
2.1.2.1 对照品溶液制备 精密称取5.00 mgD-无水葡萄糖对照品,置于50 mL量瓶中,纯化水振摇溶解,定容至刻度,即得。
2.1.2.2 供试品溶液制备 取1 mL水煎液至离心管中,加入无水乙醇适量,边加边搅拌,使其体积分数高于80%,放置过夜,滤过,残渣加热水溶解,转移至50 mL量瓶中,纯化水定容,即得。
2.1.2.3 吸光度测定 取对照品溶液、供试品溶液、纯化水(空白对照溶液)各1 mL,参照文献[4]中总多糖含量的检测方法,在486 nm波长处测定吸光度A。
2.1.2.4 线性关系考察 精密称取D-无水葡萄糖对照品溶液2、3、4、5、6、7、8、9 mL,置于10 mL量瓶中,纯化水定容至刻度,按“2.1.2.3”项下方法测定吸光度。以对照品质量浓度为横坐标(X),吸光度为纵坐标(A)进行回归,得方程为A=63.184 0X-0.004 1(r=0.999 3),在0.020 0~0.090 0 mg/mL范围内线性关系良好。
2.1.2.5 方法学考察 选择正交1号水煎液,按照2015年版《中国药典》四部指导原则进行精密度、稳定性、重复性、加样回收率试验,测得RSD均小于2.00%,平均加样回收率为101.39%,表明仪器精密度、方法重复性良好,水煎液在室温下24 h内稳定。
2.1.3 总黄酮含量测定
2.1.3.1 对照品溶液制备 精密取芦丁对照品10.00 mg至50 mL量瓶中,60%乙醇溶解并定容至刻度,即得。
2.1.3.2 供试品溶液制备 取4 mL水煎液至50 mL三角瓶中,沿壁缓缓加无水乙醇,边加边振摇,待沉淀完全后过滤,取上清液,定容至刻度,即得。
2.1.3.3 吸光度测定 同“2.1.2.3”项,参照文献[4]中总黄酮含量的检测方法,在510 nm波长处测定吸光度A。
2.1.3.4 线性关系考察 精密吸取对照品溶液2、3、4、5、6、7、8、9 mL,置于25 mL量瓶中,纯化水定容至刻度,按“2.1.3.3”项下方法测定吸光度。以对照品质量浓度为横坐标(X),吸光度为纵坐标(A)进行回归,得方程为A=11.939 0X-0.058 8(r=0.999 5),在0.016 5~0.074 2 mg/mL范围内线性关系良好。
2.1.3.5 方法学考察 选择正交1号水煎液,按照2015年版《中国药典》四部指导原则进行精密度、稳定性、重复性、加样回收率试验,测得RSD均小于1.94%,平均加样回收率为99.62%,表明仪器精密度、方法重复性良好,水煎液在室温下24 h内稳定。
2.1.4 干浸膏得率测定 取水煎液20 mL,置于已干燥恒重的蒸发皿中,95 ℃水浴蒸干后于105 ℃烘箱中干燥3 h,蒸发皿转移至干燥器内,待其冷却至室温后称定质量,计算干浸膏得率,公式为干浸膏得率=(干浸膏质量/药材总质量)×100%。
2.1.5 正交试验 依照处方配比称取相应中药饮片,选择煎煮时间(A)、加水量(B)、煎煮次数(C)作为影响因素,5-羟甲基糠醛(X1)、松果菊苷(X2)、毛蕊异黄酮葡萄糖苷(X3)、毛蕊花糖苷(X4)、总多糖(X5)、总黄酮(X6)含量及干浸膏得率(X7)作为评价指标,采用正交试验优化提取工艺[5]。因素水平见表2。

表2 提取工艺因素水平
2.1.6 数据处理 采用信息熵理论[6]确定各评价指标的权重系数,首先建立原始评价指标矩阵X,将其转换为概率矩阵P,计算各指标的信息熵(Hi)(Hi=[0.971 4 0.929 4 0.973 7 0.948 3 0.500 5 0.960 1 0.988 1])、i项指标的系数Wi(Wi=[0.039 3 0.096 8 0.036 1 0.070 9 0.685 9 0.054 8 0.016 3]),根据公式M=X1m×W1+X2m×W2+X3m×W3+…+Xnm×Wn计算综合评分M,结果见表3,方差分析见表4。由表3可知,各因素影响程度依次为C>A>B,即煎煮次数>煎煮时间>加水量;由表4可知,因素A、C有显著影响(P<0.05),而B无显著影响(P>0.05);各因素较高水平组合理论上为A3B3C3,结合生产成本考虑,最终确定为A3B3C2,即加10倍量水煎煮2次,每次2 h。

表3 正交试验设计与结果

表4 方差分析
2.1.7 验证试验 按处方配比称取相应中药饮片3份,按“2.1.6”项下优化工艺进行验证试验,结果见表5。由此可知,3批样品综合评分及各指标成分得率的RSD均小于3.00%,表明该工艺稳定可行,可用于扶正治癃方的工业化提取。

表5 提取工艺验证试验结果(n=3)
2.2 成型工艺优化
2.2.1 颗粒制备 按处方量称取相应中药饮片,按“2.1.6”项下优化工艺制备水煎液,浓缩,喷雾干燥后制得干燥恒重的浸膏粉。取相应比例的浸膏粉与辅料混合,因前者黏性较大,故以95%乙醇为润湿剂制软材(“握之成团,触之即散”),过20目筛,湿法制粒,在60 ℃烘箱中烘干整粒,即得。
2.2.2 指标测定
2.2.2.1 吸湿率 取颗粒2 g,置于已干燥恒重的扁形称量瓶中,轻摇使颗粒均匀分布于称量瓶底部,敞口放入盛有NaCl过饱和溶液的干燥器内,干燥器密封,48 h后取出称定质量,计算吸湿率[7],公式为吸湿率=[(吸湿后颗粒质量-吸湿前颗粒质量)/吸湿前颗粒质量]×100%。
2.2.2.2 成型率 取过1号筛而不过5号筛的合格颗粒,称定质量,计算成型率[8],公式为成型率=(合格颗粒质量/颗粒总质量)×100%。
2.2.2.3 溶化率 取颗粒1 g,置于已干燥至恒重的50 mL离心管中,加入100 ℃沸水20 mL,搅拌并振摇5 min,离心,弃去上清液,残渣置于80 ℃下烘干至恒重,称定质量,计算溶化率[9],公式为溶化率=(溶化的颗粒质量/颗粒总质量)×100%。
2.2.2.4 休止角 将3个漏斗串联固定在铁架台上,铁架台置于坐标纸上,保持最底端的漏斗口距坐标纸1 cm(H),将颗粒沿壁倒入最顶端漏斗口中,待坐标纸上颗粒椎体顶端触及最底端漏斗下沿时停止,记录颗粒锥体底部直径为2R,计算休止角α[10],公式为α=arctanH/R。
2.2.2.5 堆密度 将颗粒放入已干燥恒重的量筒中,反复振荡,记录体积,称定颗粒质量,计算堆密度[10],公式为堆密度=颗粒质量/颗粒体积。
2.2.3 辅料筛选 对预胶化淀粉、麦芽糊精、甘露醇、微晶纤维素进行筛选,设定浸膏粉与辅料比例为1∶1,润湿剂(95%乙醇)用量0.35倍,按“2.2.1”项下方法制备颗粒,测定其吸湿率、成型率、溶化率,结果见表6。由此可知,预胶化淀粉成型率及麦芽糊精吸湿率、溶化率明显优于其他辅料,综合考虑,选用预胶化淀粉与麦芽糊精作为辅料[11]。
2.2.4 均匀设计 选择浸膏粉与辅料比例(A)、95%乙醇用量(B)、预胶化淀粉与麦芽糊精比例(C)作为影响因素,吸湿率(X1)、成型率(X2)、溶化率(X3)、休止角(X4)、堆密度(X5)作为评价指标,G1-熵权法确定权重系数,计算综合评分(Y),采用均匀设计优化成型工艺[12-13],因素水平见表7。

表7 成型工艺因素水平
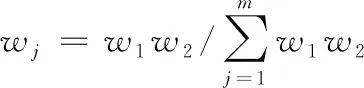

表8 评价指标权重值

表9 均匀设计结果
2.2.6 工艺验证 按“2.2.5”项下优化的成型工艺制备3批颗粒,测定吸湿率、成型率、溶化率、休止角、堆密度,计算综合评分,结果见表10。由此可知,颗粒平均综合评分高于表9最高值93.715 2分,而且各指标RSD均<3.00%,表明该工艺稳定可行。

表10 成型工艺验证试验结果(n=3)
2.2.7 临界相对湿度测定 在棕色干燥器内配置7种盐(CH3COOK、MgCl2、K2CO3、NaBr、NaCl、KCl、K2SO4)的过饱和溶液,放置48 h。取颗粒2 g,置于已干燥恒重的扁形称量瓶中,轻摇使其均匀分布在称量瓶底部,敞口放在盛有不同盐过饱和溶液的干燥器中,放置48 h后称定质量,计算吸湿率[16],结果见表11。以相对湿度为横坐标(X),吸湿率为纵坐标(Y)绘制曲线,在曲线前后拐点处做切线,结果见图2,可知前、后拐点处切线方程分别为Y=0.073 4X+0.955 4、Y=0.502 6X-26.447 4,两切线交点所对应的横坐标为临界相对湿度(CRH),其数值为63.85%。
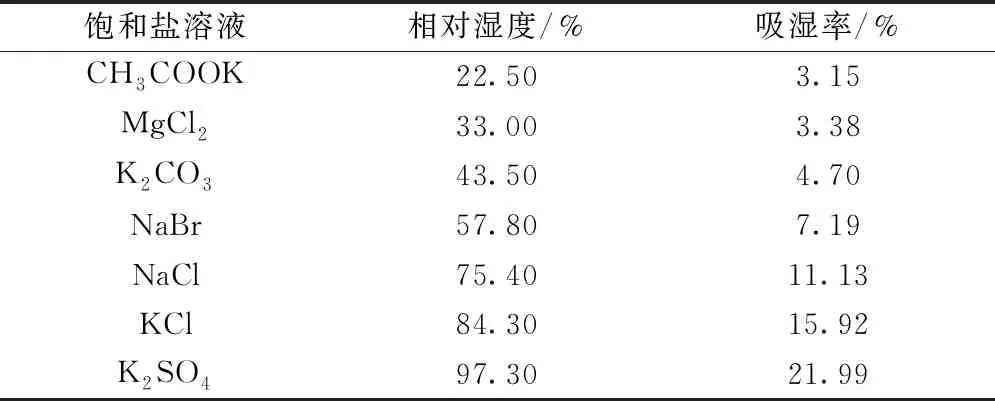
表11 颗粒吸湿率测定结果

图2 颗粒吸湿曲线
3 讨论
中药治疗过程中“多靶点、多途径”的特点与所含的多种化学成分密不可分,因此评价指标的选择是中药提取工艺研究中十分重要的环节[17]。本研究所选的7种评价指标,不仅包含毛蕊异黄酮葡萄糖苷、松果菊苷、毛蕊花糖苷等对前列腺增生有明确治疗作用的单体成分[18],又有总黄酮、总多糖等有效部位,可较全面地评价所选工艺参数的优劣。
因扶正治癃方中含熟地、肉苁蓉两味中药,故黏性大,吸湿性大,流动性差,在制成颗粒时需加入合适的润湿剂与辅料。预实验发现,当润湿剂乙醇浓度为95%时,颗粒成型性好。微晶纤维素和甘露醇作为填充剂时,所得颗粒松散、硬度不够。麦芽糊精和预胶化淀粉价格低廉、口感滑腻、具有流动性好,溶解性能好,不易吸潮等特点。两者结合使用可以很好的降低颗粒吸湿性,提高成型性与溶化性。
权重系数是中药多指标评价时必须考虑的问题之一。目前应用较多的是主观赋权法[19],这类方法虽在一定程度上体现了中药复方的配伍规律,但主观性较强,常忽略实际样本的数据信息特点。近年来,信息熵理论加权法、G1-熵权法、AHP-CRITIC混合加权法等在工艺优化中逐渐得到广泛应用。该类方法简单易行、结果精确,避免了人为确定权重的随意性和不确定性,使实验结果更加可靠、科学。
