TAA诱导的A型肝性脑病大鼠肠道通透性改变及机制研究
2021-10-26郑鋆
郑 鋆
复旦大学附属中山医院厦门医院(福建 厦门 361006)
肝性脑病(Hepatic Encephalopathy,HE)是肝脏功能严重障碍和(或)门体分流所致的中枢神经系统功能失调综合征,是急性肝衰竭和慢性肝病终末期最严重的并发症之一。按病因不同将此临床综合征分为三种类型,其中A型为急性肝衰竭相关的肝性脑病,预后最差[1]。目前,氨和肠道来源的毒素仍然是肝性脑病发病机制的核心[2],近年来研究显示,在肝硬化和慢性肝病人群中肠道通透性存在异常增高[3],间接表现为外周血内毒素水平和细菌DNA的增加;肠道屏障的破坏程度与肝功能Child分级呈正相关,在合并腹水的失代偿期肝硬化患者中肠道通透性增加更为明显[4-5]。但关于肝性脑病状态下肠道通透性改变的研究尚少,故设计了本次实验,于2020年12月至2021年3月选取雄性SD大鼠30只随机分至正常对照组和肝性脑病模型组,模型组造模成功后根据肝性脑病临床分级将其进一步细分为低级别肝性脑病组和高级别肝性脑病组;对比研究各组大鼠肠道通透性的差别并对其可能机制进行探讨,为进一步的临床研究提供基础。现报道如下。
1 材料与方法
1.1实验动物 6~8周龄雄性SD大鼠30只:购自复旦大学上海医学院实验动物中心。
1.2实验主要试剂 硫代乙酰胺(Thioacetamide,TAA)及FITC-Dextran:购自Sigma-Aldrich。
1.3实验方法
1.3.1 动物分组造模 将雄性SD大鼠随机分至正常对照组和肝性脑病模型组,正常对照组8只腹腔注射生理盐水;肝性脑病模型组22只腹腔注射TAA(300mg/kg TAA连续3天每隔24小时腹腔注射),保证大鼠的行为自由和正常的日昼时间,然后每24h进行监测以挑选出其中确实患有肝性脑病的大鼠[6],对造模成功的大鼠根据肝性脑病临床分级(表1)将其进一步细分为低级别肝性脑病组(1~2级)和高级别肝性脑病组(3~4级)。

表1 SD大鼠肝性脑病临床分级
1.3.2 离体肠道通透性测定 无菌条件下分离三组实验鼠的空肠,放置于含8.4mM HEPES,119mM NaCl,4.7mM KCl,1.2mM MgSO4,1.2mM KH2PO4,25mM NaHCO3,2.5mM CaCl2和11mM 葡萄糖的改良Krebs-Henseleit碳酸氢盐缓冲液(KHBB缓冲液)中。被分离肠段的一端用缝线结扎,然后用管饲针将100μL 异硫氰荧光素-右旋糖苷(FITC-Dextran,分子量4000,荧光剂FD-4,40mg/mL)注入肠腔,注射过程中注意避免损伤肠粘膜。肠段的另一端也用缝线结扎,形成8cm长的肠袋。用KHBB缓冲液漂洗后,将两端结扎的肠袋置入2ml KHBB液中,37℃下孵育20分钟。用荧光分光光度计测定从肠腔渗入到孵育缓冲液中的FD-4。
1.3.3 肠壁水肿程度测定 采用干湿重法比较三组实验鼠肠壁水肿程度。取空肠一小块,洗净肠内容物,普通滤纸吸干表面液体,称重(湿重),记录重量;然后放入50℃恒温培养箱中烘干,3天后再称重(干重),记录重量。根据湿、干重比,了解肠壁水肿程度。
1.3.4 紧密连接蛋白及炎症因子检测 RT-PCR法比较三组实验鼠空肠紧密连接蛋白ZO-1和Occludin及炎症因子TNF-α的表达水平。TRIzol试剂提取组织内总RNA,然后反转录为cDNA;PCR条件如下:94℃ 5分钟,之后95℃ 15秒,60℃ 30秒循环40次;内参选用β-actin,相应的引物序列如表2所示。

表2 引物序列
1.3.5 统计方法 三组数据间比较采用多因素方差分析(PCR数据采用2-△△Ct值作为原始数据参与计算),两两比较采用Bonferroni法,均为双侧检验,以P<0.05视为有统计学差异;数据分析采用SPSS 21.0,作图采用Graphpad Prism 6。
2 结果
肝性脑病模型组共获得低级别肝性脑病大鼠和高级别肝性脑病大鼠各8只,死亡6只,正常对照组8只均存活;每日的肝性脑病临床分级评估结果详见表3。观察造模结束后存活的各组动物体重、肠道通透性及相关紧密连接蛋白/炎症因子表达的变化情况如下。

表3 模型组动物评估及分组情况
2.1体重变化 实验结果提示:和对照组相比,肝性脑病组大鼠的体重有显著下降(0.06±0.04 VS-0.07±-0.02 VS-0.17±0.03,P<0.05),具有统计学差异;高级别肝性脑病组体重下降更为明显,且与低级别肝性脑病组相比也具有统计学差异(见图1)。

图1 肝性脑病组与正常对照组大鼠体重变化情况
2.2肠道通透性变化 实验结果提示:和对照组相比,肝性脑病组大鼠的单位面积离体肠道通透性(0.82±0.11 VS 1.36±0.30 VS 1.74±0.32,P<0.05)及肠壁水肿程度(3.02±0.18 VS 3.57±0.22 VS 4.44±0.43,P<0.05)均有显著增加,并具有统计学差异;高级别肝性脑病组肠道通透性增加更为明显,且与低级别肝性脑病组相比也具有统计学差异(见图2),说明肝性脑病这一病理状态下确实存在肠道屏障的破坏,且破坏程度与肝性脑病分级存在相关。

图2 肝性脑病组与正常对照组大鼠肠道通透性变化
2.3紧密连接蛋白及炎症因子表达水平变化 实验结果提示:和对照组相比,肝性脑病组大鼠的肠道紧密连接蛋白ZO-1(0.30±0.04 VS 0.27±0.05 VS 0.24±0.04,P=0.068)及Occludin(0.18±0.02 VS 0.18±0.03 VS 0.16±0.02,P=0.069)的表达下降,但不具有统计学差异,高级别肝性脑病组下降趋势更为明显,但与低级别肝性脑病组相比亦不具有统计学差异;炎症因子TNF-α表达增加(0.03±0.01VS 0.04±0.01 VS 0.08±0.02,P<0.05),具有统计学差异,高级别肝性脑病组增加更为显著,且与低级别肝性脑病组相比也具有统计学差异(见图3),说明炎症反应参与了肠漏的过程。
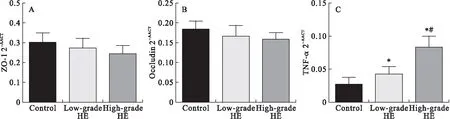
图3 肝性脑病组与正常对照组大鼠紧密连接蛋白及炎症因子表达水平变化
3 讨论
肝性脑病根据病因不同可分为A、B、C三型。在急性肝衰竭引起的A型肝性脑病中,以肝功能严重障碍因素为主,而B型肝性脑病为单纯门体旁路引起,无确切的肝细胞疾病,但临床表现与C型相同;C型肝性脑病病因则为肝硬化伴门脉高压和(或)门体分流。不同病因的HE发病机制均主要涉及三个环节:第一,循环毒素如氨的产生,其中肠道是各类毒素进入血液循环最主要的门户;第二,肝功能损伤和(或)门体分流病理生理基础存在:肝功能障碍导致肝脏的生物转化能力下降,门体旁路导致肠道毒素未经肝脏直接分流进入体循环;第三,突破血-脑屏障的毒素对在不同水平上对脑功能的损害[2]。作为毒素来源的肠道是本研究关注的核心,因在健康个体中,肠道屏障完整,具有防止肠腔内有害物质如细菌和毒素等穿过肠粘膜进入体内其他组织器官和血液循环的结构和功能;而在肝损的病理状态下,肠道屏障破坏可能导致毒素进入血液循环增加,促进HE的发生与发展。
本实验采用的是TAA致大鼠急性肝衰竭模型,该模型具有良好的重复性和可行性,是目前国内外相对成熟和常用的动物HE模型。在既往研究中观察到在造模过程中实验动物会出现不同程度的消化道症状如进食减少、明显的腹部膨隆、胃肠型等,肠壁组织病理学可见上皮细胞脱落及炎细胞浸润、绒毛坏死,符合肠道屏障功能障碍的表现[7]。这与本实验中观察到的HE状态下离体肠道通透性异常增高、肠壁水肿的结果相符合(P<0.05),且肝性脑病临床分级越高、肠道通透性增加越显著;以上都从不同角度说明肠道屏障破坏参与了HE的发病。
完整的肠道屏障包括物理屏障、化学屏障、免疫屏障、细菌屏障等,其中物理屏障主要指上皮细胞自身的完整性及细胞间的紧密连接(Tight Junction,TJ)。紧密连接包含一系列跨膜蛋白家族,包括Occludin、Claudin等,其胞内部分通过ZO蛋白与细胞内骨架结构相连接以起到锚定作用,并受饮食、激素、炎症因子等因素影响,通过磷酸化方式发生构象改变,调节肠腔内各类大分子的转运[8,9]。研究认为,在急慢性肝衰竭状态时肠道局部及全身炎症因子表达增加,而炎症反应可引起紧密连接蛋白表达水平的下调,肠道屏障异常开放,导致菌群移位、内毒素血症等,进一步加重肝损,同时肠道局部炎症状态又加剧了肠漏,形成恶性循环[10-13]。而TNF-α作为重要的促炎因子之一,已有研究提示针对肠道局部的TNF-α拮抗剂可抑制炎症反应,减轻因肠道屏障破坏引起的机体慢性炎症状态[14]。在本实验中观察到HE大鼠肠道组织局部TNF-α表达增高(0.03±0.01VS 0.04±0.01 VS 0.08±0.02,P<0.05),并具有统计学差异,紧密连接蛋白ZO-1及Occludin在基因表达水平方面存在下降趋势,与Luo等人的研究结果一致[15];说明炎症反应引起的紧密连接蛋白表达下调与肠道屏障破坏和HE的发病相关。但研究在紧密连接蛋白基因表达水平方面未得到阳性结果,可能与样本量不足及检测手段单一有关,有文献报道称,LPS诱导的肠上皮细胞损伤可导致ZO-1在蛋白表达水平上的下降(Western Blot法证实),同时免疫组化还发现其在细胞表面上的分布方式存在异常,表现为数量减少且更为碎片化[16]。
综上,肠道通透性增加是肝性脑病发生发展的重要驱动因素,而修复肠道屏障可能是未来具有吸引力的治疗手段,鉴于它能够避免针对其他一些致病机制进行治疗的不良反应(如针对肠道菌群容易产生耐药或菌群多样性减少等),并且可以和其他现有治疗手段相结合。对肠道屏障认识的不断深入,将使得未来开发修复肠道屏障的药物成为可能,以造福于临床HE患者。
