CHRND基因突变所致慢通道先天性肌无力综合征一家系
2021-10-25林慧婷张月琪张艳谭金玉曾文双
林慧婷 张月琪 张艳 谭金玉 曾文双
先天性肌无力综合征 (congenital myasthenic syndromes,CMS)是一大组由于基因缺陷导致神经肌肉接头形成、结构维持与功能异常的罕见遗传疾病[1]。其中,慢通道综合征是由于突触后膜乙酰胆碱受体(acetylcholine receptor,AChR)错义突变导致AChR离子通道持续过度开放或开放延迟的疾病,是先天性肌无力综合征中唯一呈常染色体显性遗传的疾病。本文报告1个由CHRND基因突变所致慢通道先天性肌无力综合征的家系,分析其临床表现、电生理检查所见、肌肉病理改变和基因突变及随访治疗情况。经文献检索,未见国内外相关报告,有望提高对这一特殊类型CMS的认识。
1 资料与方法
1.1 临床资料先证者男性(III 2),19岁,因“眼睑下垂、四肢无力十余年”于2019年7月就诊于香港大学深圳医院神经内科。患者自小学时期开始出现左侧眼睑下垂及四肢无力,运动后无力症状加重,休息后可缓解。上述症状缓慢进展,平素步行数百米即需休息,无法上体育课,间断出现四肢无力突然加重,表现为活动时突发无力跌倒,不能自行站起,休息后可缓解。病程无明显视物重影、呼吸困难、胸闷气促、肌肉疼痛或肌肉萎缩。既往史与家族史:胎儿发育期间正常,生长发育里程碑正常。智能发育正常。
家族中母亲(II 4)、两个舅舅(II 2,II 3)及一个舅舅的儿子(III 1)有类似病史,母亲自16岁起开始出现四肢乏力、眼睑下垂、眼球活动障碍。两个舅舅及其中一个舅舅的儿子表现为四肢乏力,运动后加重,上三层楼需休息。自诉外婆有无力症状,具体不详。否认近亲结婚。先证者家系图见图1。
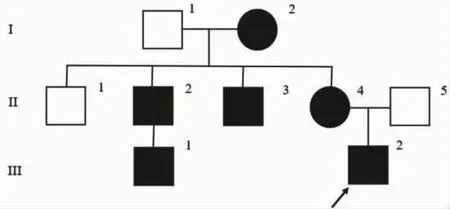
图1 家系图表示先证者(III 2)。
1.2 体格检查发育正常,体型正常。心肺查体无异常。四肢关节及脊柱未见畸形。神清言利。高级皮层功能未见明确异常。左侧眼睑下垂,覆盖瞳孔3―9点钟位,双眼外展受限,留白2 mm,余颅神经查体未见明确异常。颈屈肌肌力4级,卧位屈颈抬头困难。步态呈鸭步。四肢肌容积正常。四肢近端肌力4级,远端肌力 5+级。四肢深浅感觉对称,腱反射对称,共济稳准。四肢病理征阴性,脑膜刺激征阴性。自主神经功能未见明显异常。疲劳试验:左眼自发阳性,右眼上视10 s内出现眼睑下垂。上肢平举疲劳试验40 s,下肢蹲起疲劳试验小于30 s,均呈阳性。
1.3 辅助检查血、尿、便常规正常。肝肾功能、血糖、血脂、电解质、心肌酶谱、甲状腺功能、自身免疫抗体谱、感染筛查均正常。静息乳酸为2.2 mmol/L。心电图示窦性心律。超声心动图、双眼眼底照相、肺功能检查均未见异常。入院后行新斯的明试验为阴性。行乳酸最小运动量试验为阴性(运动前乳酸为2.2 mmol/L,运动后即刻乳酸为3.2 mmol/L,运动后10 min乳酸为2.3 mmol/L)。重症肌无力抗体检测:抗AChR抗体、肌肉特异性酪氨酸激酶抗体、人低密度脂蛋白受体相关蛋白4抗体均为阴性。
神经电生理检查:行四肢运动神经传导(motor conduction velocity,MCV)检测,运动传导的潜伏期、波幅、传导速度未见明显异常。四肢感觉神经传导(sensory conduction velocity,SCV)未见异常。右侧三角肌、胫前肌、股内侧肌针极肌电图静息相未见自发电位,小力收缩时运动单位电位(motor unit potential,MUP)平均时限变窄,波幅稍低,呈轻度慢性肌源性损害改变。双侧面神经 (眼轮匝肌)、双侧副神经(斜方肌)、双侧尺神经(小指展肌)低频(2、3、5 Hz)重复电刺激可见波幅递减。双侧尺神经高频重复电刺激(30、50 Hz)未见波幅递减或递增。
1.4 肌肉活检病理检查经患者同意,取其左侧三角肌行组织活检。普通光镜HE染色下可见组织肌间纤维结缔组织无明显增生。肌纤维大小大致均匀,直径40~60 μm,未见明显萎缩肌纤维。未见核聚集。偶见个别内核肌纤维。未见嗜酸性高收缩肌纤维,未见坏死、再生肌纤维。组织内未见明显炎细胞浸润。余组织学染色、酶组织化学染色、免疫组织化学染色未见明确病理性改变(图2)。电镜超微病理未见明确病理性改变。
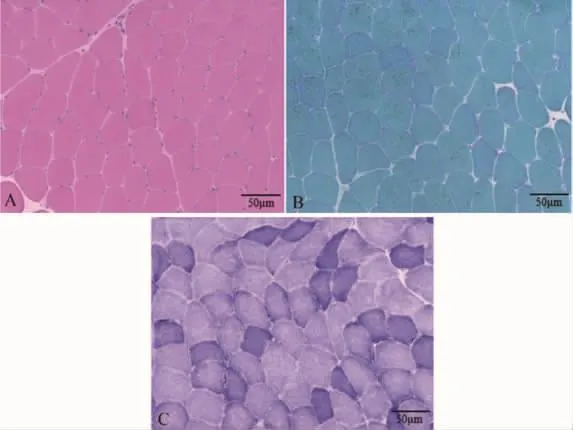
图2 左侧三角肌活组织病理 A为HE染色(20×),B为MGT染色(20×),C 为 NADH 染色(20×)。
1.5 基因检测先证者(III 2)二代测序显示CHRND基因c.826G>A杂合突变,为错义突变,突变位点来源于母亲(II 4),父亲该位点无突变。有相似症状的一个舅舅(II 2)及其儿子(III 1)一代测序示相同位点突变(图3)。该位点未有文献报告,经MutationTaster、SIFT、Polyphen-2 预测软件分析提示有害。

图3 CHRND基因突变所致先天性肌无力综合征家系全外显子测序结果 图中红色箭头指示突变位点c.826G>A。在这个家系中,先证者及其母亲、舅舅、舅舅的儿子存在c.826G>A的杂合突变,其父亲无突变。
1.6 治疗结合患者家系表现为常染色体显性遗传及基因检测结果,诊断慢通道型先天性肌无力综合征。予患者、患者母亲、舅舅及其儿子氟西汀20 mg每天一次口服治疗。患者服药2周后肌无力症状有改善,但仍有活动不耐受,加量至40 mg后肌无力症状显著改善,目前眼睑无明显下垂,活动耐力较前增加,步态接近正常。患者舅舅及其儿子服药后四肢乏力症状改善明显。患者母亲服药后感心律不齐,减量至10 mg后心脏不适症状缓解,肌无力轻微改善。
2 讨论
先天性肌无力综合征是一组由基因突变导致神经肌肉接头功能异常的遗传性疾病。编码神经肌肉接头的突触前膜、突触间隙和突触后膜众多蛋白的基因出现影响功能的致病突变,都可能导致CMS[1-3]。其发病率约1/50万~1/20万。至今已发现可导致CMS的致病基因达30余种,包括 CHRNE、CHRNAL、CHRNBL、CHRND、RAPSN、COLQ、DOK7、MUSK、CHAT、GFPTl、AGRN、SCN4A等[3-4]。
CMS常表现为常染色体隐性遗传,仅慢通道综合征表现为常染色体显性遗传。慢通道综合征由ENGLE于1982年首次报告[5],是由于乙酰胆碱受体错义突变导致AChR离子通道持续过度开放导致临床症状发生。AChR通道持续过度开放可引起肌肉细胞内钙超载,造成终板肌病,使终板皱折减少,突触间隙增宽,结合区皱折变性,AChR丧失,接头胞质内细胞器变性,细胞核凋亡。同时,由于突触形态的改变,AChR的丢失,延长的终板电位在通道活动期间因时间上的蓄积造成去极化阻滞,使得神经肌肉信号传导的正常状态受损[6]。本例患者检测到CHRND基因突变,位于2q37.1,主要编码乙酰胆碱受体的δ亚基,为AChR5个亚基之一,突变可导致AChR表达减少及功能异常,临床上常见的表型有慢通道先天性肌无力综合征3A 型(slow-channel congenital myasthenic syndrome type 3A,SCCMS-3 A)、3 B 型、3 C 型与乙酰胆碱受体缺乏症[7-8]。
CMS常在新生儿期或幼年开始出现波动性眼外肌、肢体肌肉或球部肌肉无力,易疲劳,可出现脊柱侧弯畸形,心肌和平滑肌通常不受累,病情进展十分缓慢,但可出现突然加重。少数患者表现为18岁以后发病的成年晚发型,临床表现具有高度异质性[1,3]。
由于在电生理检查上均可表现为低频重复神经电刺激CMAP波幅递减,临床上均可表现为疲劳现象、运动不耐受,CMS需与重症肌无力相鉴别。而血清学指标在鉴别诊断中十分重要。CMS的诊断需要求所有与重症肌无力相关抗体均为阴性。本例患者重症肌无力相关抗体均阴性,支持CMS诊断。对于慢通道综合征的患者,药物试验阴性(新斯的明试验或溴吡斯的明)无效亦可作为鉴别诊断的要点之一。此外,CMS合并阳性家族史时还需与代谢性肌病(如线粒体肌病)或其他先天性肌病相鉴别,血清乳酸、乳酸运动试验等可作为初筛检查之一,肌肉组织病理检查为鉴别上述疾病的重要指标[3]。
CMS的治疗主要根据其分型及基因突变的类型决定。溴吡斯的明、沙丁胺醇、麻黄碱、奎尼丁与氟西汀等可通过不同的作用机制治疗不同类型CMS。对于慢通道综合征,首选药物为氟西汀和奎尼丁,其主要通过阻滞开放的通道及缩短通道开放的时间起到治疗目的。使用奎尼丁需注意其心律相关的副作用(加重房室传导阻滞、延长QT间期等),需动态监测心电图及药物血清浓度,氟西汀因安全性更高在临床上使用更多[9-10]。
先天性肌无力综合征多为儿童期起病,以波动性肌无力、运动易疲劳等为主要表现,低频刺激下波幅递减可作为诊断线索。多数罕见病缺乏有效的治疗方法,而CMS是少数可用药物治疗及改善临床症状的罕见病。对于这一异质性罕见疾病需从临床、电生理、肌肉病理、分子基因多方面结合进行精准诊断,从而更好指导临床用药。
