SPE/GC-MS测定3种水产品中的喹哪啶残留
2021-10-20徐闪浪黄和高平陈日檬曾丹丹陈营寿
徐闪浪 ,黄和 *,高平*,陈日檬,曾丹丹,陈营寿
1. 广东海洋大学食品科技学院(湛江 524088);2. 广东省水产品加工与安全重点实验室(湛江 524088);3. 水产品深加工广东普通高等学校重点实验室(湛江 524088);4. 广东海洋大学分析测试中心(湛江 524088);5. 湛江市食品药品检验所(湛江 524022)
渔用麻醉剂是通过抑制活的水产品中枢神经系统来缓解其应激反应,降低运输途中的伤亡率,减少经济损失目的的药物,在国内外被广泛应用于水产品的捕捉、运输、取样、测量等渔业生产和科学研究的过程中[1-2]。喹哪啶(又称2-甲基喹啉)是一种无色油状的喹啉类化合物,广泛应用于医药、染料制备等方面[3]。喹哪啶可用于鱼类的短时间麻醉操作辅助活鱼运输。相比较于其他渔用麻醉剂,喹哪啶快速诱导、恢复迅速,且价格低廉,成为新兴的渔用麻醉剂之一[4-5]。但喹哪啶具有一定毒性,对小鼠的LD50为1 230 mg/kg[6],对人的皮肤、眼睛、黏膜、上呼吸道具有刺激性作用,此外有试验研究表明喹哪啶对人体具有慢性致突变作用[7]。倘若其残留在水产品中,可能对人体健康造成一定危害。对于渔用麻醉剂的管理,国内缺乏有效的使用标准[8],为避免喹哪啶在水产品生产和流通过程中泛滥使用,对水产品中喹哪啶麻醉剂残留进行准确高效的检测具有重要的实际意义。
有关喹哪啶检测方法报道相对较少,主要有紫外分光光度(UV)法、气相色谱(GC)法[9]和液相色谱-串联质谱(LC-MS/MS)法[10-11]。UV法灵敏度较低,定性定量不准,不适合用于痕量分析;GC法虽然操作简单,普及性较广,但容易受到复杂基质的干扰,从而影响目标物的准确定性与定量;LC-MS/MS法抗干扰能力强,灵敏度高,定性准确,但仪器昂贵。以南美白对虾、石斑鱼和鳜鱼为试验对象,采用气相色谱-质谱法[12-14]结合固相萃取法[15-16],建立水产品中喹哪啶麻醉剂残留的检测方法,为水产品中喹哪啶残留的测定提供新的检测手段。
1 材料与方法
1.1 试剂与仪器
喹哪啶(CAS号91-63-4,纯度≥98%,上海麦克林生化科技有限公司);乙腈、正己烷、乙酸乙酯(均为HPLC级,美国Honeywell公司);固相萃取柱:乙二胺-N-丙基柱(PSA,500 mg,6 mL)、氨基柱(NH2,500 mg,6 mL)、弗罗里硅土柱(Florisil,1 000 mg,6 mL)、硅胶柱(Silica,1 000 mg,6 mL,艾杰尔科技有限公司);苯基柱(Phenyl,500 mg,6 mL,美国Agilent公司);石墨化炭黑柱(GCB,500 mg,6 mL,美国Supelco公司);均质子(1 cm×2 cm,美国Agilent公司)
GCMS-QP2010 Ultra气相色谱-质谱联用仪,配有电子轰击离子源(EI)(日本SHIMADZU公司);RV10旋转蒸发仪(德国IKA公司);Centrisartg-16C高速冷冻离心机(赛多利斯科学仪器(北京)有限公司);Vac Elut 20固相萃取装置(美国Agilent公司);Milli-Q IQ7000超纯水机(德国Merck公司)。
1.2 试验方法
1.2.1 标准溶液的配制
准确称取适量喹哪啶标准品(精确至0.01 mg)用乙酸乙酯溶解、定容,配制成质量浓度1.0 mg/mL储备液,于-18 ℃避光保存备用。使用时用乙酸乙酯稀释成质量浓度10,20,50,100,200和500 μg/L的系列标准工作液,临用现配。
1.2.2 样品前处理
将石斑鱼、鳜鱼、南美白对虾等样品取肌肉部分,用匀浆机绞碎混匀,于-18 ℃以下条件冷冻保存备用,使用时取出自然解冻 。
提取:称取2 g(精确至0.01 g)样品置于50 mL离心管中,加入2 mL超纯水和1颗均质子,旋涡振荡1 min,加入10 mL乙腈,旋涡振荡5 min后超声提取10 min,加入2 g氯化钠(NaCl)手摇1 min,以5 000 r/min离心10 min,上清液转移至另1支50 mL离心管中;加入10 mL乙腈重复提取1次,合并乙腈提取液,加入2 g无水硫酸镁(MgSO4),剧烈振摇1 min,以5 000 r/min离心5 min,上清液转移至50 mL鸡心瓶中,40 ℃旋转蒸发至近干,3 mL正己烷复溶待净化。
净化:依次使用5 mL乙酸乙酯、5 mL正己烷活化Silica固相萃取柱,将复溶液全部转移至固相萃取柱中,用3 mL正己烷润洗鸡心瓶,全部倒入固相萃取柱中,依靠重力自然流出。用5 mL正己烷淋洗固相萃取柱,弃掉全部流出液,用5 mL乙酸乙酯洗脱并收集,40 ℃水浴氮吹至体积小于2 mL,再用乙酸乙酯定容至2 mL供GC-MS分析。
1.2.3 气相色谱-质谱条件
1) 色谱条件
色谱柱:DB-17MS毛细管柱(30 m×0.25 mm×0.25 μm);进样口温度230 ℃。升温程序:初始柱温50 ℃,保持2 min,以10 ℃/min升至200 ℃,保持2 min,以20 ℃/min升至280 ℃保持17 min。载气,高纯氦气(纯度≥99.999%);总流量35 mL/min;恒压模式90 kPa;进样模式,不分流模式;进样量1 μL。
2) 质谱条件
离子源温度230 ℃;接口温度260 ℃;电离方式,电子轰击源(EI);电离能量70 eV;溶剂延迟5 min;检测方式,选择离子监测(SIM)模式。在上述条件下喹哪啶标准溶液总离子流图见图1,喹哪啶保留时间、定量和定性离子及丰度比见表1。

表1 喹哪啶的保留时间以及定量、定性离子
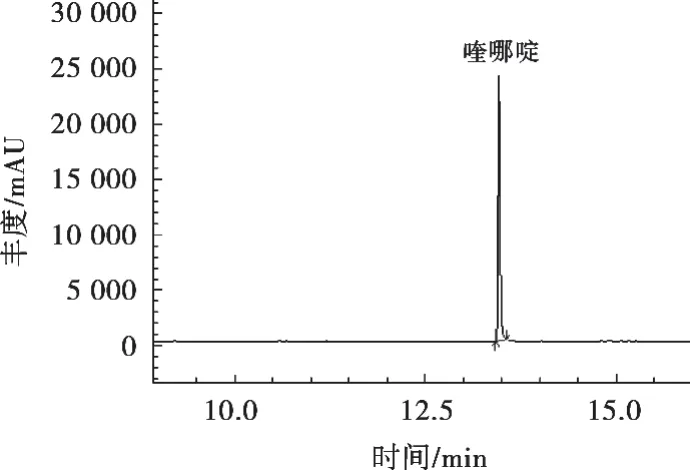
图1 喹哪啶标准溶液SIM模式下总离子流图(50 μg/L)
2 结果与讨论
2.1 分析条件选择
2.1.1 色谱柱的选择
试验考察SH-RTx-5MS、DB-5MS、DB-17MS这3种色谱柱对喹哪啶的分离效果(见图2)。采用SHRTx-5MS色谱柱时,目标物出峰时间较早,噪音较大,基线不稳,峰形较差,拖尾严重;采用DB-5MS色谱柱时,出峰时间稍晚,虽基线和峰形均有所改善,但仍有拖尾现象;采用DB-17MS色谱柱时,目标物质出峰时间较理想,分离效果好,基线平稳,峰形尖锐。因此,选择DB-17MS色谱柱为分析柱。
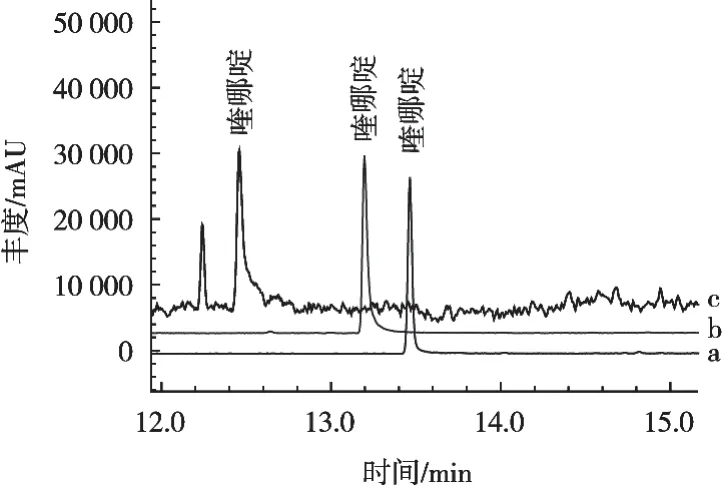
图2 不同色谱柱对喹哪啶分离效果的影响
2.2 前处理条件优化
2.2.1 提取溶剂的选择
试验选择乙腈、乙腈+乙酸乙酯(1∶1,V/V)、正己烷+丙酮(3∶1,V/V)、正己烷4种有机溶剂作为提取溶剂,考察其对水产品中喹哪啶的提取效果。称取2 g水产品(石斑鱼)空白样品于50 mL离心管中,添加250 μg/kg喹哪啶标准溶液,分别使用4种溶剂进行提取,提取液定容至20 mL,上机测定(总离子流见图3)并计算回收率,每种提取溶剂设置3组平行试验。

图3 不同提取溶剂对喹哪啶的提取效果
结果显示,正己烷+丙酮(3∶1,V/V)和正己烷作为提取溶剂时,虽回收率均≥120%,但杂质较多,后期净化难度大,对仪器和色谱柱影响和伤害也较大。乙腈和乙腈+乙酸乙酯(1∶1,V/V)作为提取溶剂时,回收率为95%~100%,提取效果无明显差别,但乙腈+乙酸乙酯(1∶1,V/V)作为提取溶剂时,回收率不稳定,因此试验选择乙腈作为提取溶剂。在提取时加入均质子辅助,振荡过程中进一步混匀样品,增大提取效果。同时在提取溶液中加入NaCl促进水相和有机相分离,不仅减轻后续净化过程的除水压力,还增大待测组分在乙腈相中的分配比,提取效果更好。
2.2.2 固相萃取柱的选择
水产品样品基质较复杂,提取液需经净化处理才可上机测定。固相萃取法是试验室最为常用的净化方式,其操作简单,净化效果较好[17]。试验称取2 g水产品(石斑鱼)空白样品中,加入50 μg/kg喹哪啶标准溶液,按照1.2.2进行提取后,分别拟采用NH2柱、Florisil柱、Silica柱、PSA柱、Phenyl柱、GCB柱[18-19]6种固相萃取柱净化喹哪啶提取液,上机测定并计算和比较加标回收率。每种固相萃取柱设置3组平行试验,不同固相萃取柱净化后的回收率的比较见图4。
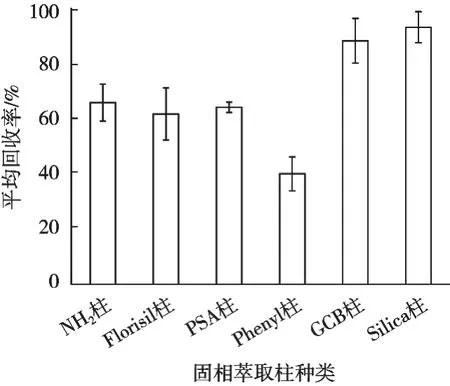
图4 不同类型固相萃取柱对喹哪啶加标回收率的影响
6种不同固相萃取柱的回收率差异较大,其中NH2柱、Florisil柱、PSA柱、Phenyl柱4种柱子净化效果略差,回收率均≤70%;GCB柱和Silica柱2种柱子净化效果较好,回收率均≥88%。但GCB柱净化时用乙腈-甲苯混合洗脱,甲苯毒性大,氮吹时间较长。综合考虑试验成本、试剂毒性、试验时长等多方面因素,故最终选择Silica柱作为净化固相萃取柱。
2.2.3 洗脱溶剂体积的选择
考察乙酸乙酯作为洗脱溶剂时,洗脱溶剂体积对喹哪啶回收率的影响。选择3,4,5,6,7和8 mL的6组不同洗脱溶剂体积,每组进行3次重复试验。结果显示,洗脱溶剂体积3 mL时,回收率为55%~65%;随着洗脱溶剂体积增加,回收率有所提高,洗脱溶剂体积5 mL时,回收率为88%~95%;但洗脱溶剂体积>5 mL时,回收率没有明显增加。因此,考虑到节约成本和避免水浴氮吹时间太长,试验选择5 mL乙酸乙酯作为洗脱溶剂。
2.3 方法线性范围、检出限和定量限
将1.2.1配制的质量浓度分别为10,20,50,100,200和500 μg/L系列标准溶液按照1.2.3条件进行分析,以标准溶液的质量浓度(x,μg/L)为横坐标,以峰的面积(y)为纵坐标,建立线性回归方程为y=745.54x+3 324.08。结果表明,喹哪啶在10~500 μg/L范围内,线性关系良好,相关系数为0.999 5。根据相关文献[20],以3倍信噪比(S/N≥3)计算方法检出限(LOD),以10倍信噪比(S/N≥10)计算方法定量限(LOQ),同时结合仪器实际响应情况,计算得喹哪啶的方法检出限为5 μg/kg,定量限为20 μg/kg。
2.4 加标回收率和精密度
空白南美白对虾、石斑鱼、鳜鱼肌肉样品中分别添加20,40和200 μg/kg喹哪啶标准溶液,每个浓度设置6个平行同时设置1个空白对照,按照1.2.2进行提取、净化、上机测定,计算喹哪啶的加标回收率及相对标准偏差(SRSD),结果见表2。结果显示,喹哪啶平均加标回收率为80.0%~98.2%;相对标准偏差(SRSD)为3.3%~11.5%,表明试验方法回收率较好,重复性高,符合残留分析要求[21]。
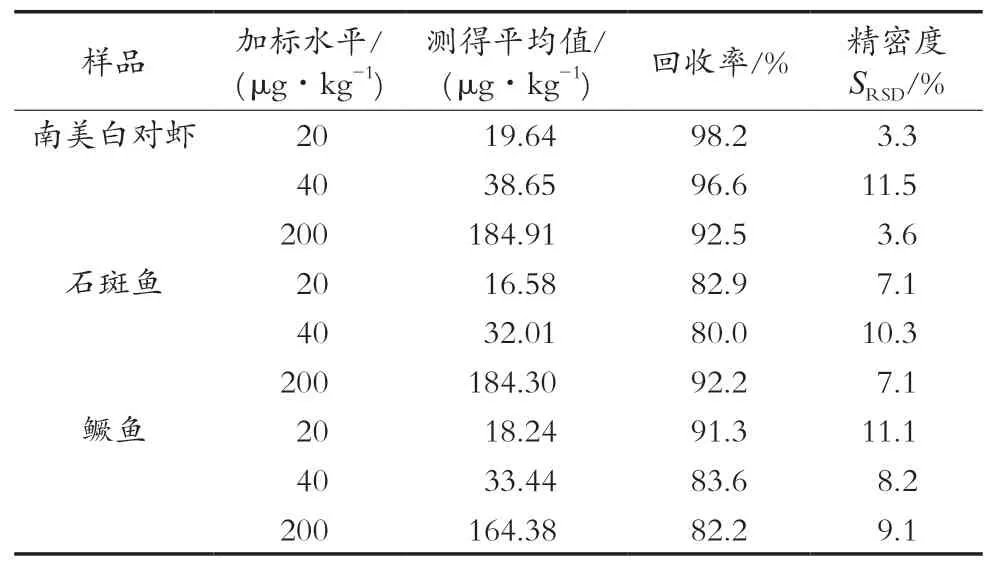
表2 加标样品的回收率和精密度(n=6)
空白石斑鱼样品和石斑鱼加标样品SIM模式下总离子流图见图5。

图5 空白和加标石斑鱼在SIM模式下总离子流图
2.5 实际样品测定
采用建立的方法对湛江某水产品批发市场随机购买的10份水产品样品进行检测,结果显示所有样品均未检出喹哪啶。
3 结论
建立气相色谱-质谱联用仪测定水产品中喹哪啶麻醉剂残留的检测方法,其检出限为5 μg/kg,定量限为20 μg/kg,平均回收率为80.0%~98.2%;相对标准偏差为3.3%~11.5%。该方法简单快捷,检测成本低,回收率高,重现性好,适合用于水产品中喹哪啶麻醉剂残留的测定。
