气相色谱-串联质谱法测定沙坦类制剂中N-亚硝基二甲胺和N-亚硝基二乙胺含量
2021-10-20马斐田海燕刘向凌肖宗阳
马斐,田海燕,刘向凌,肖宗阳
(德州市食品药品检验检测中心,山东 德州 253000)
世界卫生组织国际癌症研究机构致癌物清单显示,N-亚硝基二甲胺(NDMA)和N-亚硝基二乙胺(NDEA)均属于2A类致癌物质。ICH M7指南中也明确指出该类化合物具有较高致癌性[1]。近年来,随着药品监管的不断深入,在沙坦类原料药中发现含有上述两种杂质,而且部分原料药中NDMA含量超出欧盟标准200余倍,严重威胁患者的生命健康。因此,各原料药公司已经将NDMA和NDEA列入内部质量控制标准中。截至目前,国内外已建立多种原料药和制剂的NDMA和NDEA含量检测方法[2-11],包括利用气相色谱-质谱法进行缬沙坦及厄贝沙坦原料药内NDMA和NDEA含量检测,利用超高效液相色谱-静电场轨道阱高分辨质谱检测方法进行法莫替丁原料药及制剂中NDMA含量检测[11]等。成品药物中NDMA及NDEA的检验与原料药质量控制相互补充,不可或缺,但仍未见沙坦类药物制剂中NDMA及NDEA检测方法的报道。
本研究利用气相色谱-串联质谱(GC-MS/MS)对缬沙坦、厄贝沙坦、替米沙坦、坎地沙坦4种药物的不同剂型(片剂、胶囊、分散片)进行检测,建立了沙坦类成药制剂中NDMA及NDEA的GC-MS/MS检测方法;对30批次沙坦类制剂进行检测,发现1批阳性样品,NDMA含量低于限量值,NDEA未检出。本方法对于服务药品监管,构筑药品安全最后一道防线具有积极的促进作用。
1 实验部分
1.1 仪器、试剂与材料 气相色谱-串联质谱联用仪7890B+7010B(美国Agilent公司);精密电子天平XSE105(瑞士Mettler-Toledo公司);试管涡旋振荡器[德国海道尔夫(Heidolph)公司];H1850R台式高速冷冻离心机(长沙高新技术产业开发区湘仪离心机仪器有限公司); 13 mm×0.22 μm PTFE滤膜(上海安谱实验室科技股份有限公司)。
NDMA对照品(2 000 μg·mL-1,纯度100%,BW901305-2000-A,批号:A1909201,北京坛墨质检科技有限公司);NDEA对照品(100 μg·mL-1,纯度100%,BW902298-100-A,批号:A1911043,北京坛墨质检科技有限公司);二氯甲烷(色谱纯,科密欧公司)。
1.2 对照品及供试品溶液制备
1.2.1 对照品溶液 精密量取NDMA标准溶液0.5 mL至100 mL容量瓶中,用二氯甲烷溶解并稀释至刻度,制备浓度为10 μg·mL-1的NDMA对照品溶液。分别精密量取NDMA对照品溶液(10 μg·mL-1)5 mL、NDEA对照品溶液(100 μg·mL-1)0.5 mL至同一200 mL量瓶中,加二氯甲烷溶解并稀释至刻度,制成含NDMA、NDEA均为250 ng·mL-1的对照品储备液。分别精密量取上述储备液适量,用二氯甲烷稀释,制备成质量浓度为0、2.5、5、10、30、60、125、250 ng·mL-1的系列标准工作溶液。
1.2.2 样品处理 精密称取各品种细粉适量(约相当于主成分100 mg),置于15 mL离心管中,加入二氯甲烷5 mL,2 000 r·min-1涡旋混匀3 min,4 ℃条件下10 000 r·min-1离心10 min,上清液经0.22 μm PTFE滤膜滤过,待进样分析。
1.3 色谱条件 毛细管柱(型号Agilent VF-WAX ms,30 m×0.25 mm,0.25 μm),升温程序:初始柱温40 ℃,保持0.5 min,20 ℃·min-1的速率升至200 ℃,保持8 min。进样口温度:250 ℃,载气为氦气,流速1 mL·min-1,不分流进样,进样量1 μL。
1.4 质谱条件 采用EI源,电压为70 eV,接口温度为280 ℃,离子源温度为230 ℃,增益因子10,溶剂延迟3.5 min,多反应监测检测模式见表1。

表1 多反应监测检测模式参数
2 结果与讨论
2.1 试验条件的考察
2.1.1 仪器选择及质谱条件优化 根据Carcinogenicity Potency Database(CPDB)数据库中致癌物质的TD50值来计算每日可接受的摄入量(ADI),NDMA、NDEA TD50值分别为0.095 9和0.026 5 mg·(kg·d)-1[12-13],按照人体重50 kg来计算NDMA的每天可接受摄入量为:0.095 9 mg·(kg·d)-1×50 kg/50 000 =96 ng·d-1,此时对应肿瘤发生风险为十万分之一。结合药品说明书及FDA公告中查阅到缬沙坦、厄贝沙坦、替米沙坦、坎地沙坦的每日最大用药量分别为320、300、80、32 mg[13]。限度=ADI/每日最大用药量(以缬沙坦为例,NDMA 0.3 ppm,NDEA 0.08 ppm)。根据供试品溶液中沙坦类的质量浓度为0.02 g·mL-1,可见NDMA及NDEA的定量下限须低于3 ng·mL-1、0.8 ng·mL-1(即限度浓度的50%)。国内外的检测方法现有HPLC-UV、GC-MS[1-3]、GC-MS/MS[4-7]、LC-MS/MS、LC-HRMS等,有数据表明串联质谱灵敏度比单四级杆质谱要高10倍,直接进样法比顶空灵敏度更高近1倍,为排除假阳性,满足灵敏度要求,也鉴于目前串联质谱仪的普及情况,选择GC-MS/MS直接进样法和UPLC-MS/MS两种方法进行测试。分别对质谱条件进行优化,对于目标物给予不同碰撞能量,找出信噪比高的离子对,GC-MS/MS质谱条件见表1。
2.1.2 色谱条件优化 分别考察UPLC-MS/MS和GC-MS/MS分析检测NDMA的灵敏度,发现GC-MS/MS的检出限更低,因此选用GC-MS/MS进行检测。有文献报道[14],使用非极性色谱柱会造成亚硝胺色谱峰拖尾,使用中等、强极性色谱柱亚硝胺色谱峰尖锐对称,因此选用专为极性化合物设计的Agilent VF-WAX ms柱进行分析,能实现更高的灵敏度。
2.1.3 样品前处理 对比取样量相当于主成分500 mg和100 mg,分别进行加标回收,阳性样品测定,阳性结果均一致,加标回收率、灵敏度均满足要求,取样量较大导致辅料太多,提取液比较黏稠,杂质较多,对色谱柱及质谱污染较大,所以选择约相当于主成分100 mg的取样量进行测定。
NDMA和NDEA在甲醇和二氯甲烷中均有较好的溶解性,因此分别采用甲醇和二氯甲烷对制剂进行提取,加标量0.005 μg·kg-1,提取溶剂体积均为5 mL,比较回收率,结果表明二氯甲烷的回收率及精密度较甲醇更好,因此采用二氯甲烷作为提取溶剂。
提取时间不足也是影响目标物回收率的重要因素,超声效果较好,但是容易产热,可能造成NDMA的产生[15],且二氯甲烷易挥发,因此选择涡旋提取,并要合理控制提取时间。设置1、3、10 min 3个提取时间,对比胶囊、分散片2种不同剂型5 ng·mL-1加标浓度下的平均回收率为78%、95%、93%,结果表明,提取时间为3 min时,可满足检验要求。
2.2 方法学验证
2.2.1 专属性 将溶剂(二氯甲烷)、对照品溶液、阴性样品溶液、阴性样品加标分别进样测定,相同保留时间处,溶剂及阴性样品均无干扰(见图1~3)。
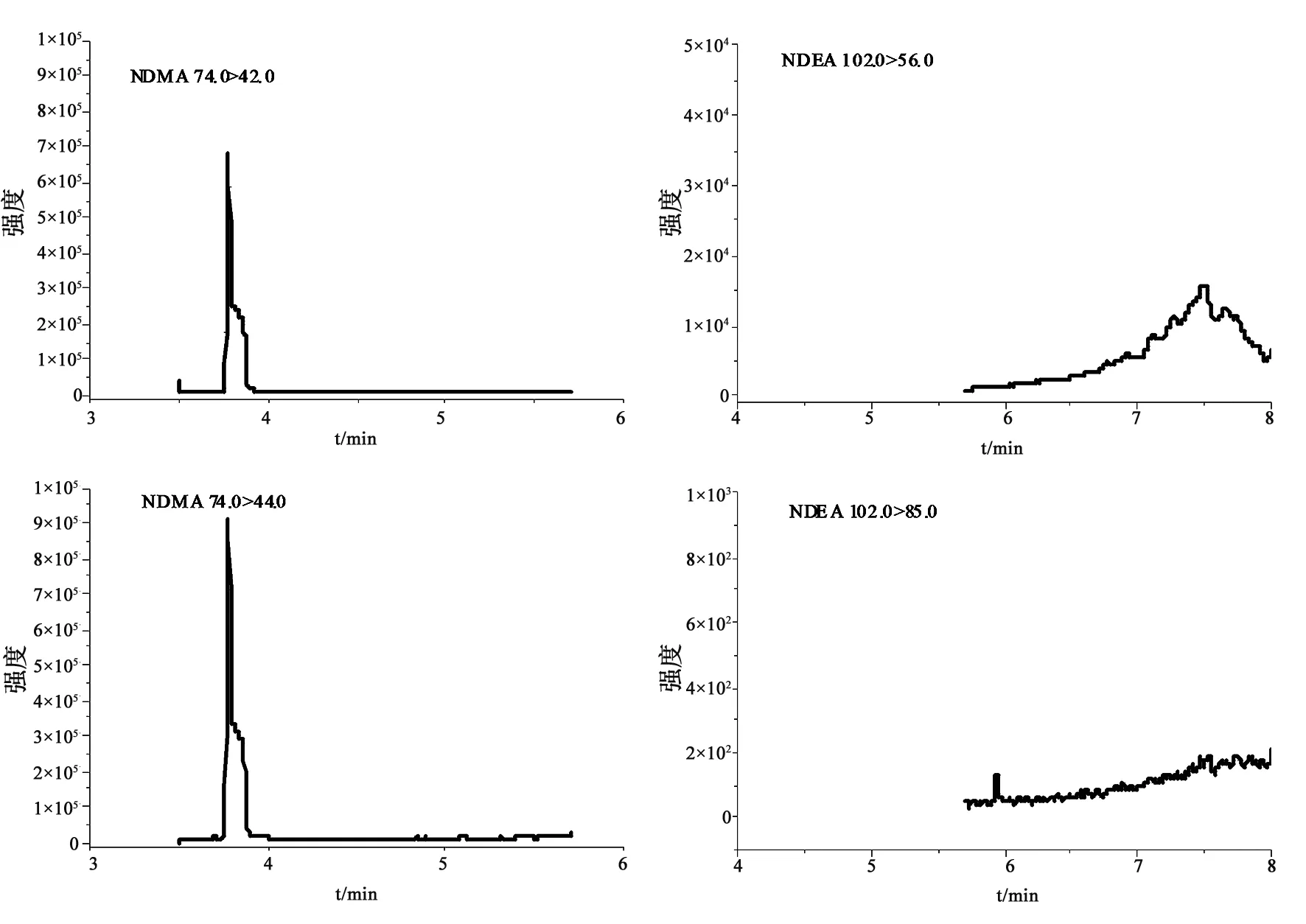
图1 溶剂(二氯甲烷)提取离子色谱图

图2 NDMA、NDEA标准溶液提取离子色谱图
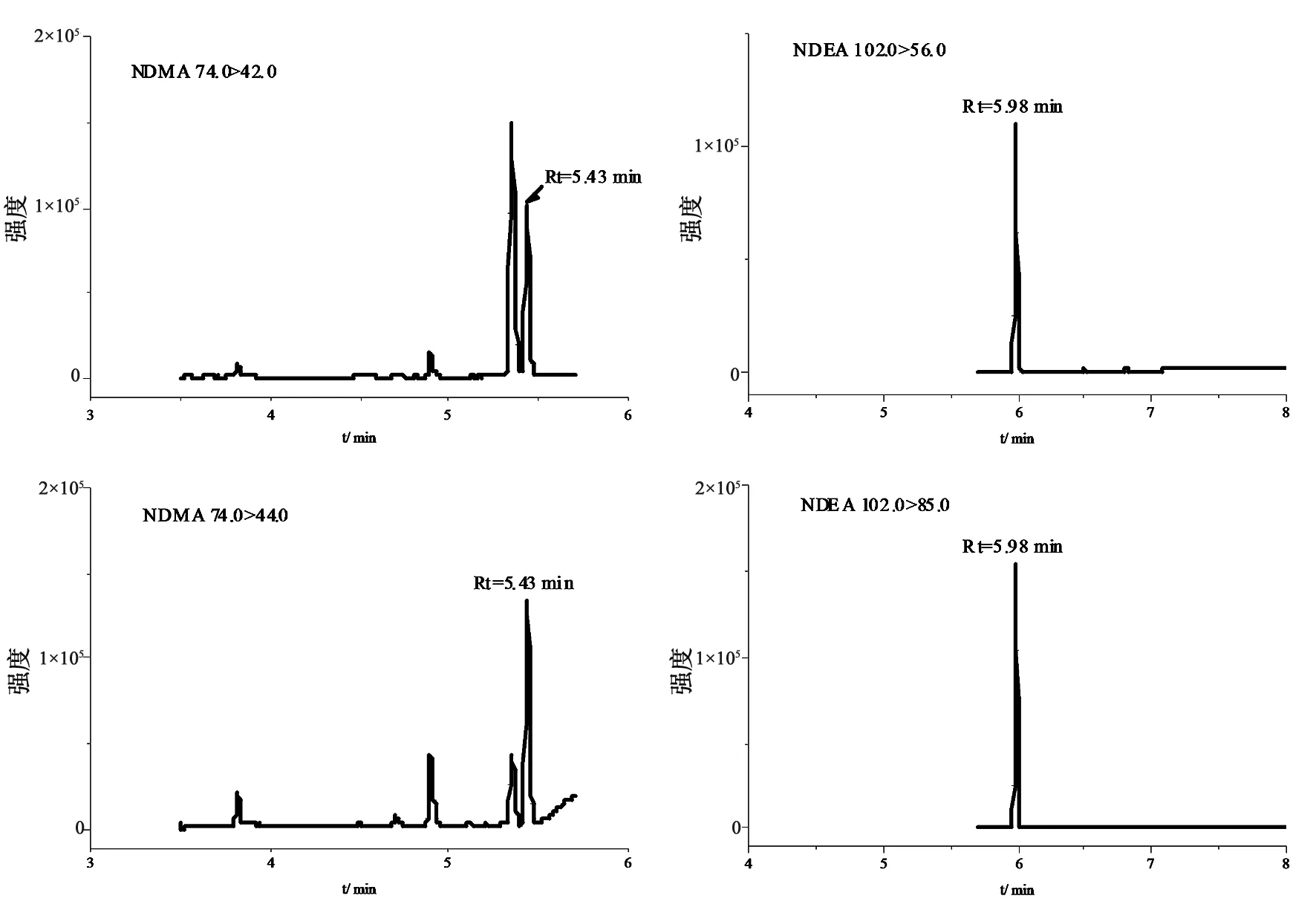
图3 加标样品提取离子色谱图
2.2.2 基质效应 选取缬沙坦分散片、厄贝沙坦分散片、替米沙坦胶囊的空白样品,分别配制低(5 ng·mL-1)、中(10 ng·mL-1)、高(30 ng·mL-1)3个水平的空白基质加标溶液和二氯甲烷溶剂标准溶液,分别进样测定,计算两种溶液中NDMA、NDEA响应值的百分比。结果表明,NDMA、NDEA在3种不同基质中的基质效应分别为90.1%~108.7%、92.6%~103.5%、95.1%~101.3%,表明基质效应不影响检测。
2.2.3 线性范围 将“1.2.1”项下配制的系列浓度对照品溶液进样测定。以所得峰面积为纵坐标Y,标准溶液的质量浓度为横坐标(X,ng·mL-1),建立线性回归方程,并计算相关系数(r)。结果表明,NDMA在0~250 ng·mL-1范围内,线性良好,回归方程为:Y=7 733.9X+1 443.1,r=0.999 7。NDEA在0~250 ng·mL-1范围内,线性良好,回归方程为:Y=4 024.1X-1 292.5,r=0.999 7。
2.2.4 检出限和定量限 将1 ng·mL-1的NDMA和NDEA标准溶液用二氯甲烷定量逐级稀释后进行测定,以信噪比S/N≥3作为检出限,以信噪比S/N≥10作为定量限。结果表明方法的检出限NDMA为0.1 ng·mL-1,NDEA为0.08 ng·mL-1,定量限NDMA为0.3 ng·mL-1,NDEA为0.2 ng·mL-1。
2.2.5 回收率与精密度 按照“1.2.2”项下精密称取缬沙坦分散片、替米沙坦胶囊、厄贝沙坦分散片适量,至15 mL离心管中,各精密加入250 ng·mL-1的混合标准溶液适量,制备成低中高3个浓度的加标回收样品各6份,依照本方法处理,进样测定,计算平均回收率,结果见表2。NDMA平均回收率为86.9%~113.2%,RSD为2.7%~7.4%;NDEA平均回收率为91.1%~105.3%,RSD为2.8%~8.4%,表明回收率及重复性均良好。

表2 加标回收率及相对标准偏差
2.2.5 稳定性与耐用性 将5 ng·mL-1的NDMA和NDEA空白样品加标溶液,按照试验条件放置0、1、2、4、8、12 h后进样测定,记录色谱图和峰面积。结果显示NDMA和NDEA的峰面积的RSD为7.5%和8.9%,说明该条件下进行样品测定12 h内稳定性良好。
改用相似固定相的柱子Agilent DB-WAX(0.25 mm×30 m,0.25 μm)进行分析,NDMA和NDEA的检出限、分离度均能符合要求,表明方法耐用性良好。
2.3 样品测定 应用所建立的方法对30批沙坦类制剂样品进行检测,根据《国家药典委关于缬沙坦国家标准修订稿》[15]中规定NDMA不得过0.3 ppm、NDEA不得过0.08 ppm。测得阳性样品1批,其计算公式为:
[式中m0为规格80 mg,ω为平均片重或装量0.160 2 g,m为取样量0.367 3 g,V为定容体积5 mL,ω为含量(ppm)],结果如表3所示。

表3 阳性样品测定结果
3 结论
本研究建立了沙坦类制剂中NDMA、NDEA的GC-MS/MS检测方法,其中,NDMA、NDEA的线性范围为0~250 ng·mL-1,方法的检出限NDMA为0.1 ng·mL-1、NDEA为0.08 ng·mL-1,定量限NDMA为0.3 ng·mL-1、 NDEA为0.2 ng·mL-1。NDMA平均回收率为86.9%~113.2%,RSD为2.7%~7.4%;NDEA平均回收率为91.1%~105.3%。RSD为2.8%~8.4%。应用该法对30批样品进行检测,检出阳性样品1批,NDMA含量低于限量,NDEA未检出。本方法灵敏度高、专属性好、回收率高、线性范围宽,操作简便、快速,已用于沙坦类制剂的检验,填补了该检验技术空白。
