先天性神经母细胞瘤六例临床病理分析
2021-10-18张艳丽胡俊波郭鹏刘涵瀚
张艳丽 胡俊波 郭鹏 刘涵瀚
神经母细胞瘤(neuroblastoma,NB)是交感神经系统的胚性肿瘤,起源于肾上腺髓质或椎旁神经节的交感细胞,发生于胎儿或产后早期。先天性神经母细胞瘤是新生儿期最常见的颅外恶性肿瘤之一,占所有先天性肿瘤的20%。在所有活产新生儿中,发病率约1~10/10万[1]。肿瘤多原发于腹部,尤其是肾上腺髓质,其次还有纵隔、颈部等。神经母细胞瘤具有广泛的肿瘤异质性,可发生肿瘤的自发消退,或分化为良性的神经胶质瘤等,也可迅速恶化、转移。本文回顾性分析6例先天性神经母细胞瘤,并进行临床病理特征、免疫组化染色观察及相关文献复习,探讨其临床、病理学特点、发病机制及生物学行为、预后等,旨在提高对该肿瘤的认识及鉴别诊断,并深入了解该肿瘤的治疗与预后。
对象与方法
一、对象
我院2016年1月~2020年11月术后病理诊断为先天性神经母细胞瘤的病例6例,其中4例为先天性囊性神经母细胞瘤。所有病例切片均由2位高年资病理医师复阅确认。6例患儿中,男性4例,女性2例。5例于孕晚期常规超声检查发现胎儿腹腔内包块,发现时胎龄为33周、32周、30周、34周、36周,平均胎龄33周;1例为出生后10天因黄疸、新生儿肺炎入院查体发现后纵隔肿块;患儿手术治疗时的年龄分别为1天、11天、1天、1个月、5天、2个月。6例病人的临床资料见表1。

表1 6例先天性NB的临床及病理资料
二、方法
6例均经手术单纯肿瘤切除,术前、术后均未行放疗、化疗,切除标本行病理检查。所有标本均经4%中性缓冲甲醛固定,梯度脱水,石蜡包埋、4 μm厚切片,HE染色和光镜下观察。免疫组化染色采用EnVision二步法,罗氏全自动免疫组化仪进行染色,并用已知阳性切片作阳性对照。一抗包括PHOX2B、 Syn、 CgA、CD56、CD99、FLI-1、Ki-67等,抗体及试剂盒均购自Dako公司。荧光原位杂交(FISH)检测采用N-myc探针,试剂盒购自康录生物技术股份有限公司,均按试剂说明书进行操作。
结果
1.巨检:6例标本均经手术完整切除。2例肉眼可见包膜;肿瘤最大直径3.5~6 cm,平均约4.5 cm;4例切面为囊性,质软,色灰黄灰白,部分区可见出血;2例切面为实性,均质、质软,色灰白,鱼肉样,可见出血。5例发生部位为肾上腺,1例为后纵隔。
2.镜检:低倍镜下,4例为囊性,囊壁外层可见肾上腺组织,肿瘤局限于肾上腺内(图1);6例均见小圆形原始肿瘤细胞由纤细的不完整纤维分割成巢团状(图2)、不规则分布,部分区可见出血、变性,2例可见钙化。高倍镜下,肿瘤细胞小而规则,稍大于淋巴细胞,胞浆稀少,边界欠清晰,核圆形,深染,染色质细腻,可有小核仁(图3);1例局灶区肿瘤细胞排列呈假菊形团样结构(图4),可见极少量神经毡;6例肿瘤细胞均未见典型节细胞分化,分型为未分化型,有丝分裂-核碎裂指数(mitosis-karyorrhexis index,MKI)为低度(<100/5 000)。
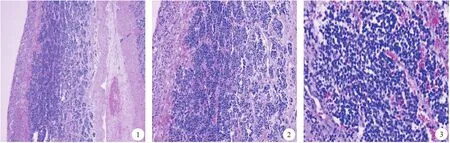
图1 肿瘤细胞局限于肾上腺组织内(HE×40) 图2 肿瘤细胞由纤细的纤维分隔成巢状(HE×100) 图3 肿瘤细胞小而规则,胞浆稀少,核圆形,深染,染色质细腻,可有小核仁(HE×200)

图4 肿瘤细胞排列呈假菊形团样结构(HE×200) 图5 肿瘤细胞PHOX2B染色(+)(Envision二步法×100) 图6 肿瘤细胞CD56染色(+)(Envision二步法×100)
3.免疫组化及免疫荧光原位杂交:6例PHOX2B阳性(图5),6例Syn阳性,6例CgA阳性,6例CD56阳性(图6),1例CD99局灶阳性,3例FLI-1阳性,4例Ki-67增殖指数>40%,2例Ki-67增殖指数<5%。免疫荧光原位杂交:6例N-MYC检测均显示N-MYC未扩增。
4.随访:随访截止2020年11月,1例失随访,5例患儿获取随访资料,随访时间1~53个月;5例均无病生存(分别术后53个月、14个月、11个月、6个月、6个月),暂无复发、转移。
讨论
NB是一种未分化的胚胎神经细胞瘤,最常发生于肾上腺(90%),也可见于后纵隔或交感神经节。先天性NB少见,且NB通常为实性,很少出现囊性变,故腹膜后的囊性NB更是极为罕见。先天性NB通常发现于妊娠晚期,以32周以后较为多见,诊断时平均胎龄约36周左右[2]。
切除标本肉眼观通常较大,平均直径4.5 cm。早期出血多时类似血肿,也可发生囊性变,外观如肾上腺囊肿。镜下观:肿瘤细胞呈大小一致的小圆形原始未分化细胞,常由纤细的不完整的纤维分隔呈模糊的结节状,可形成假血管样或腺泡状结构,常见出血、坏死及钙化灶。钙化可能为突出特征,可表现为粉末样嗜碱性点彩状,同心圆或围绕单个肿瘤细胞呈鸡笼样结构。肿瘤细胞小而规则、核圆形、深染,胞浆少,边界不清。1/4~1/3可见Homer-Wright菊形团。电镜特点:可见有纵行排列微小管的外周齿状突起,其特点是含有儿茶酚氨的电子致密的小圆颗粒。依据2003年修订的Shimada分类系统[3],其组织学分类分为4个亚型:(1)神经母细胞瘤:未分化型、分化差型、分化型;(2)节细胞神经母细胞瘤(混杂型);(3)节细胞神经母细胞瘤(结节型):经典型、多结节型、巨结节型、无结节型;(4)节细胞神经瘤:即将成熟型和成熟型。1999年Shimada根据神经母细胞瘤分化程度、基质多少、年龄、MKI等,将NB分为预后良好型、预后不良型。本文报道的6例先天性NB组织学病理特征与以往报道的NB相同,未发现特殊的组织学特征。6例均为未分化型,其中4例为先天性囊性NB。免疫表型:表达神经元标志物(CD56、NSE和NF)、神经内分泌标志物(CgA和Syn)和交感肾上腺标志物(酪氨酸羟化酶、NB84和GD2)等,但辅助诊断的敏感性和特异性较差。PHOX2B阳性是诊断神经母细胞瘤具有高度敏感性和特异性的的免疫标志物,其在所有未分化型和分化型神经母细胞瘤中均可见弥漫的细胞核表达。 本文报道的6例形态学及免疫表型与文献报道相符。
预后相关因素及分子机制包括以下几个方面[3-5]:(1)年龄及确诊时的分期;(2)组织学类型;(3)儿茶酚氨及其代谢产物的水平;(5)TrK家族;(6)染色体核型(DNA倍性);(7)N-MYC扩增;(8)染色体大片段丢失或获得。
先天性NB根据临床生物学行为不同,转归差异较大,可自然退化,或分化为良性的神经胶质瘤等,也可迅速恶化、转移。相关研究证实,胎儿期NB的生物学特征与新生儿期NB有很大不同,其发现年龄早,分期多为Ⅰ、Ⅱ、Ⅳ-S期,一般无N-MYC扩增和双倍体DNA表达,常与1号染色体断臂远端(1p36.1-p36.2或1p34-p35)缺失相关,多数病理检查为未分化型,部分可自行消退,预后常常良好[6]。先天性NB的诊断时间及诊断时的肿瘤分期被公认为其预后的独立因素,5年无病生存率及存活率分别为91%和96%。胎儿NB退化的时间有较大差异性,常发生在诊断后1~8个月内,34%患儿可发生在诊断后1年内。因此有部分报道认为对胎儿NB无需过度治疗,每月进行超声检查、密切随访等待可能的自发消退、若肿瘤增大或不消退再进行单纯手术治疗或手术后化疗即可,但这一观点仍存在争议。本文报道5例为胎儿期发现肿物,1例为新生儿期发现肿物,分期5例为Ⅰ期,1例为Ⅳ-S期,病理组织学均为未分化型,N-MYC均未发现扩增,未行DNA、1p17q等染色体检查。6例均在出生后不久行单纯的肿块切除,未行术后放化疗等。目前除1例失随访外,其余5例均无病生存,但大多数病例随访时间较短,需要进一步密切长期随访。
