高通量虚拟筛选CDK2/Cyclin A2靶点抑制剂
2021-10-15叶成濠李恩民许丽艳陈广慧
叶成濠,梁 亨,李恩民,许丽艳,李 鹏,陈广慧
(1.汕头大学理学院化学系,汕头515063;2.汕头大学医学院基础医学研究所,汕头515041;3.香港中文大学(深圳)生命与健康科学学院,深圳518172)
细胞周期与肿瘤的关系密切,当细胞周期的调控机制受到破坏时,正常细胞存在向肿瘤细胞转化的潜在风险[1~3].细胞周期蛋白依赖性激酶(CDKs)隶属于丝氨酸/苏氨酸蛋白激酶,目前已发现13个家族成员,其中CDK2在真核细胞周期调控中发挥关键作用,与癌症发生及发展密切相关[4],因此发现靶向CDK2的抑制剂成为研究热点[5~7].
当CDK2与Cyclin A2以复合蛋白形式存在时,其催化活性被激活,调控细胞G1期到S期的转变[8].研究发现,CDK2/Cyclin A2复合蛋白在乳腺癌、口腔癌、食管鳞状细胞癌异常高表达[9,10].Kontopidis等[11]通过对比CDK2/抑制剂和CDK2/Cyclin A2复合物/抑制剂的结构,发现CDK2/Cyclin A2的ATP结合位点中Lys33和Asp145残基发生0.1~0.2 nm的位移,如图S1(本文支持信息)所示,使复合蛋白ATP区域结合口袋发生改变,导致针对CDK2的抑制剂失效,说明CDK2和CDK2/Cyclin A2蛋白是两个不同的靶点.因此,精准靶向CDK2/Cyclin A2复合蛋白的抑制剂才能更好地抑制激活后的CDK2.目前,实验上已有针对CDK2/Cyclin A2复合蛋白的药物研究,如NU6271[12]和N-&-N1[13]分子对复合蛋白表现出良好的抑制效果,半抑制浓度(IC50)分别为45和43 nmol/L;Bettayeb等[14]融合了Meridianin和Variolins两种化合物的结构特征,得到了针对复合蛋白具有较强抑制作用的化合物Meriolins,IC50为11 nmol/L.但由于上述抑制剂存在选择性差、溶解度低及毒性大等缺点,至今尚无应用于临床的靶向CDK2/Cyclin A2的小分子药物.
本文采用高通量虚拟筛选的方法,旨在发现针对CDK2/Cyclin A2复合蛋白的小分子抑制剂.基于DrugBank[15],ChEMBL[16]及TCM@Taiwan[17]3个数据库近90万种小分子进行筛选,采用药效团模型、药物体外药代动力学(ADME)计算、分子对接、机器学习、聚类、毒性预测以及分子动力学模拟方法,发现对复合蛋白具有抑制作用的先导化合物,采用质心分析、自由能形貌图、MM/PBSA和平均非共价作用(aNCI)分析药物-蛋白的作用模式,筛选流程图如Scheme 1所示.
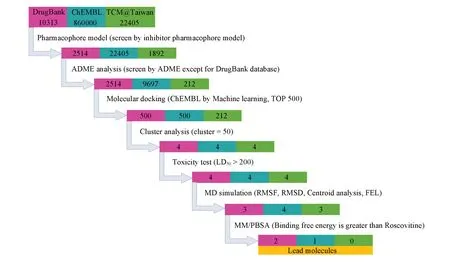
Scheme 1 Workflow of high⁃throughput screening of lead compounds on CDK2/Cyclin A2 target
1 计算方法
1.1 虚拟筛选
CDK2单体蛋白(PDB ID:2A4L)[18]和CDK2/Cyclin A2复合物蛋白(PDB ID:3DDQ)[19]从PDB Bank中获得,采用Swiss SPDBV 4.10[20]对蛋白质晶体结构进行预处理;候选分子来自DrugBank(10313)[15],ChEMBL(860000)[16]和TCM@Taiwan(22405)[17]数据库,如Scheme 1所示.首先,基于实验研究的10种靶向CDK2/Cyclin A2复合蛋白的抑制剂构建药效团模型,针对不同数据库进行筛选,并采用MOE 2015.2程序包[21]完成.
其次,根据Lipinski规则将ChEMBL[16]和TCM@Taiwan[17]数据库中筛选得到的苗头化合物(Hit compound)进行ADME分析[22].ADME是指药物在体内经过吸收、分布、代谢和排泄的药代动力学过程.筛选规则包括分子量≤500,氢键供体数目≤5,氮和氧原子数目总数≤10以及分子的脂水分配系数(MolLogP)≤4.15.
再采用机器学习(Machine Learning,ML)模型,以DrugBank数据库分子与复合蛋白的对接打分数据作为训练集,预测ADME筛选后的ChEMBL数据的分子与靶点的对接打分.通过比较训练集和验证集的决定系数R2来评估模型效果.将预测打分高的分子采用高精度的MMFF94力场进行优化并转为pdbqt格式的文件,并与靶点进行分子对接,采用AutoDock Vina程序[23]完成.
然后,采用Rdkit程序[24]建立机器学习聚类模型,处理由3个不同数据库筛选得到的分子.目的是使簇内药物相似度最大,簇与簇之间差异最小.聚类中心簇数目设置为50,以分子谱图作为特征,采用K-Means方法进行聚类.
最后,对筛选分子的毒性进行评估.通过ProTox-Ⅱ服务器(http://tox.charite.de/protox_ii)[25,26]进行,该服务器基于机器学习的方法,采用33种模型定量和定性地评估化合物的致突变性、致癌性、半数致死量(LD50)值和免疫毒性.
1.2 分子动力学模拟
为了验证虚拟筛选得到的苗头化合物与蛋白质的结合稳定性,对体系进行分子动力学(MD)模拟.首先,采用GAFF力场描述筛选得到的药物分子[27,28],在B3LYP泛函和6-31G(d,p)基组的水平上,计算配体分子的约束静电势(RESP),拟合出原子电荷,采用Amber99 sb-ILDN力场描述蛋白质[29];随后,以蛋白质-配体复合物为中心构建一个0.8 nm的TIP3P水模型盒子并设置周期性边界条件,为了保持体系的电中性,使用钠离子和氯离子代替部分水分子以产生0.15 mol/L NaCl的溶剂盒子;然后,为防止在平衡模拟过程中发生剧烈重排,对体系进行能量极小化计算以及100 ps的限制性动力学模拟;最后,采用恒温恒压(NPT)系综,模拟温度为310 K,压强为1.0×105Pa,对体系进行50 ns的MD模拟.能量极小化以及MD模拟过程中均通过LINCS算法对所有氢键进行限制,并采用GROMACS 2018.4程序[30]完成.
基于MD模拟的轨迹进行均方根波动(RMSF)、均方根位移(RMSD)和回旋半径(Rg)分析.RMSF能揭示蛋白质的各残基在模拟期间的平均位置的波动信息,采用下式计算:

式中:xi为原子的位置;T为时间间隔;tj为特定的时刻;x为该原子的平均位置.
RMSD可以说明靶蛋白和配体之间结合的稳定性,计算公式如下:

式中:di为原子的位置;N为系统中原子的数量.
Rg值用于表征分子结合的紧密程度,可以采用下式计算:

式中:r为原子与其质心的距离;m为一个原子的质量.Rg越小,说明配体与靶点结合得越紧密.
对于分子的RMSD、Rg值和吉布斯自由能3个变量,采用GROMACS和xpm2txt.py脚本绘制三维自由能形貌图(Free energy landscape,FEL),其能量最低点代表体系的稳定构象.
1.3 MM/PBSA结合自由能计算及能量分解
采用MM/PBSA方法计算蛋白与配体的结合自由能[31,32].截取最后10 ns MD轨迹,每隔100 ps选1帧,共100帧进行结合自由能和能量分解计算.采用g_mmpbsa程序[33]计算MM-PBSA结合自由能的具体公式如下:
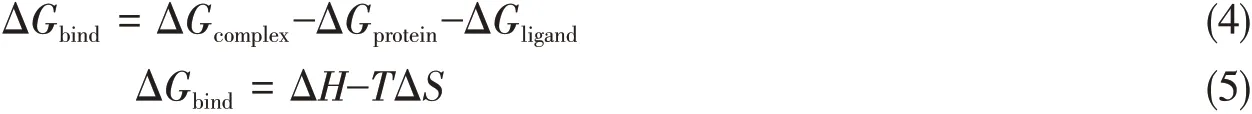
式中:ΔGbind为结合自由能;TΔS为定温下复合物的熵变;ΔH为体系的焓变,可通过下式表示:

式中:ΔEMM为复合物在气相下的焓变;ΔGsol为溶解自由能.将式(6)代入式(5)中得到下式:

由于配体与蛋白质的结合过程中熵变一般不大,在计算结合自由能的过程中可以忽略.因此式(7)可以简化为下式:

式中:ΔEMM为van der Waals作用(ΔEvdW)和静电作用(ΔEele)的总和,如下式所示:

式(8)中,ΔGsol等于极性组分溶剂化能(ΔGPB,sol)和非极性组分溶剂化能(ΔGnonpolar,sol)的总和.其中极性部分对自由能的贡献ΔGPB,sol采用Poisson-Boltzmann(PB)方法计算;而非极性自由能的贡献ΔGnonpolar,sol通过分子可及表面积(SA)计算:

最后,将式(9)和式(10)代入式(8)中,得到下式:
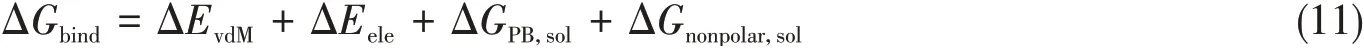
可见,采用MM/PBSA方法最终将结合自由能分解为以上4个部分.
1.4 平均非共价作用分析
aNCI分析是基于分子密度计算约化密度梯度函数(RDG),从而描述配体与蛋白质之间的弱相互作用[34,35].以MD模拟的最后一帧轨迹作为起始构象,在相同条件下进行1 ns的MD模拟,每隔2 ps取一帧共得到501帧.由所得构象的分子密度得到平均密度(------ρ(r))和平均密度梯度(--------∇ρ(r)),通过式(9)计算平均约化密度梯度(aRDG)[35]:

采用Multiwfn 3.8程序[36]对蛋白质配体复合物进行aNCI分析,通过等值面的形式揭示配体与蛋白的作用机制,明确弱相互作用区域.
2 结果与讨论
2.1 CDK2/Cyclin A2抑制剂的虚拟筛选
选取已报道的10个靶向CDK2/Cyclin A2复合蛋白,半抑制浓度在2~15600 nmol/L的抑制剂构建药效团模型[13,14,19,37~41],如表S1(本文支持信息)所示.以RMSD为0.07 nm作为阈值,对DrugBank(10313),ChEMBL(860000)和TCM@taiwan(22405)3个数据库进行虚拟筛选,分别得到2514,22405和1892个苗头化合物.
采用Rdkit程序[24]通过Lipinski’s规则对分子化合物的ADME性质进行评估,再进一步筛选化合物.发现ChEMBL和TCM@taiwan数据库中符合药效团模型的分子中分别有9697和212个满足ADME特征.由于DrugBank数据库为FDA认证的数据库,因此2514个分子被认为全部符合ADME要求.
将DrugBank和TCM@Taiwan数据库中2514和212个分子分别与CDK2/Cyclin A2靶点对接.以DrugBank数据库中分子与靶点的对接数据建立机器学习模型,预测ChEMBL数据库中9697个分子与靶点的对接打分.以数据库中化合物的理化性质作为特征,将DrugBank的数据以8∶2分成训练集和验证集.分别对随机森林[42]、神经网络[43]和XGBoost[44]3种回归模型的预测效果进行评估,结果如表S2(本文支持信息)所示.发现XGBoost模型效果最佳,在训练集和验证集中的R2分别为0.97和0.93,如图1所示.故采用该模型预测ChEMBL top 500分子的打分并进行对接验证,发现其对接打分绝对值均在19.1以上,其中有95个分子在23.9以上,如表1所示.
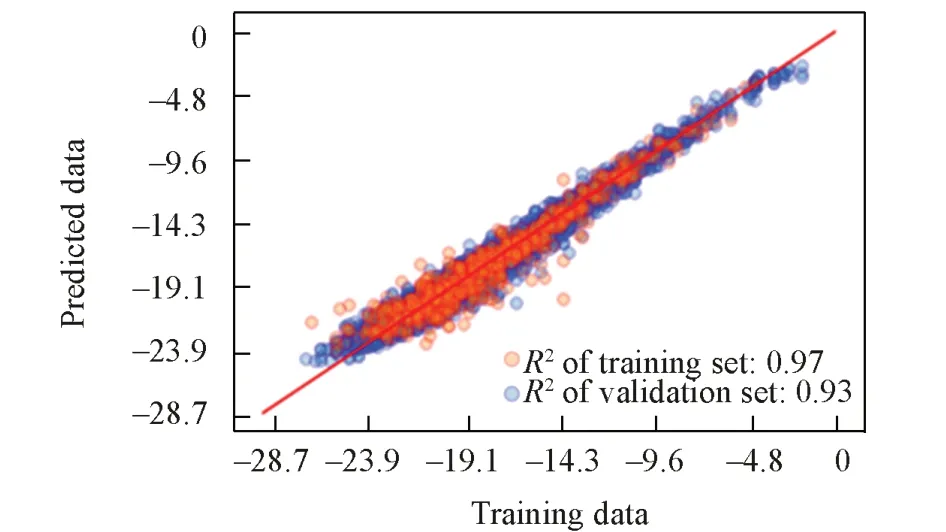
Fig.1 Scatter plots of predicted and validated docking scores between molecules of DrugBank database and CDK2/Cyclin A2 target based on XGBoost machine learning model
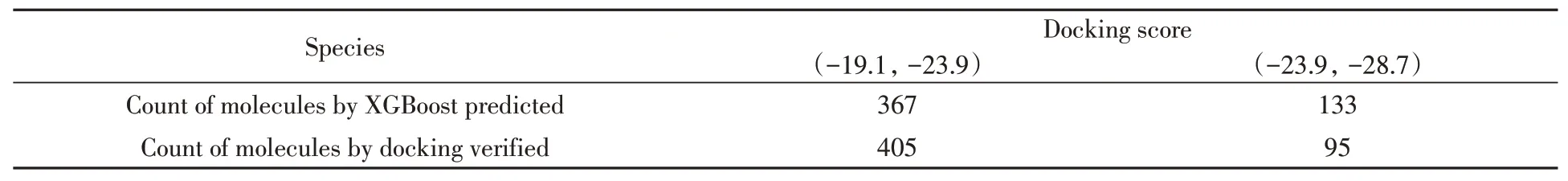
Table 1 Total counts of top 500 molecules predicted by XGBoost model and the number verified by docking in ChEMBL database
为了确保筛选得到的化合物具有化学多样性和代表性,将每个数据库对接打分前10%的分子进行聚类分析.根据Morgan指纹[45]将分子划分成50个簇并将模型可视化,如图S2(本文支持信息)所示.DrugBank,ChEMBL和TCM@Taiwan数据库分子的结构最大聚类距离分别为35,14和70,差距较大.因此,将3个数据库分子分别聚类.选取每个数据库中对接打分绝对值大于25.1的top 4苗头化合物共12个.发现属于DrugBank数据库的4个分子的官能团种类较少,结构相对简单;ChEMBL数据库中的4个分子含有羟基、氰基、环丙基等取代基;TCM@Taiwan数据库中的4个分子含有复杂的母环结构,且官能团的数目较多,表现出较大的结构多样性.对12个苗头化合物进行毒性预测,结果(表S3,本文支持信息)表明,全部分子的LD50在200~1500 mg/kg区间内,属于低毒性到中等毒性范围.
2.2 小分子与CDK2/Cyclin A2复合物的分子动力学模拟验证
对这12个分子以及阳性药Roscovitine与靶点形成的13个复合物体系分别进行50 ns的MD模拟,通过RMSF、RMSD、质心分析以及自由能形貌图(FEL)评估结合稳定性,分别如图2、图S3和图S4(本文支持信息)所示.由图2(A)~(C)可见,这12个复合物的RMSF与阳性药相近,介于0.05~0.5 nm之间.由图2(D)~(F)可见,DrugBank-8482和TCM-37233的RMSD较大,分别为1.0和0.8 nm;而其余10个分子的RMSD值与阳性药Roscovitine分子大致相同,位于0.15~0.5 nm,且均已经达到平衡状态.说明蛋白能与这10个配体形成稳定的复合物.
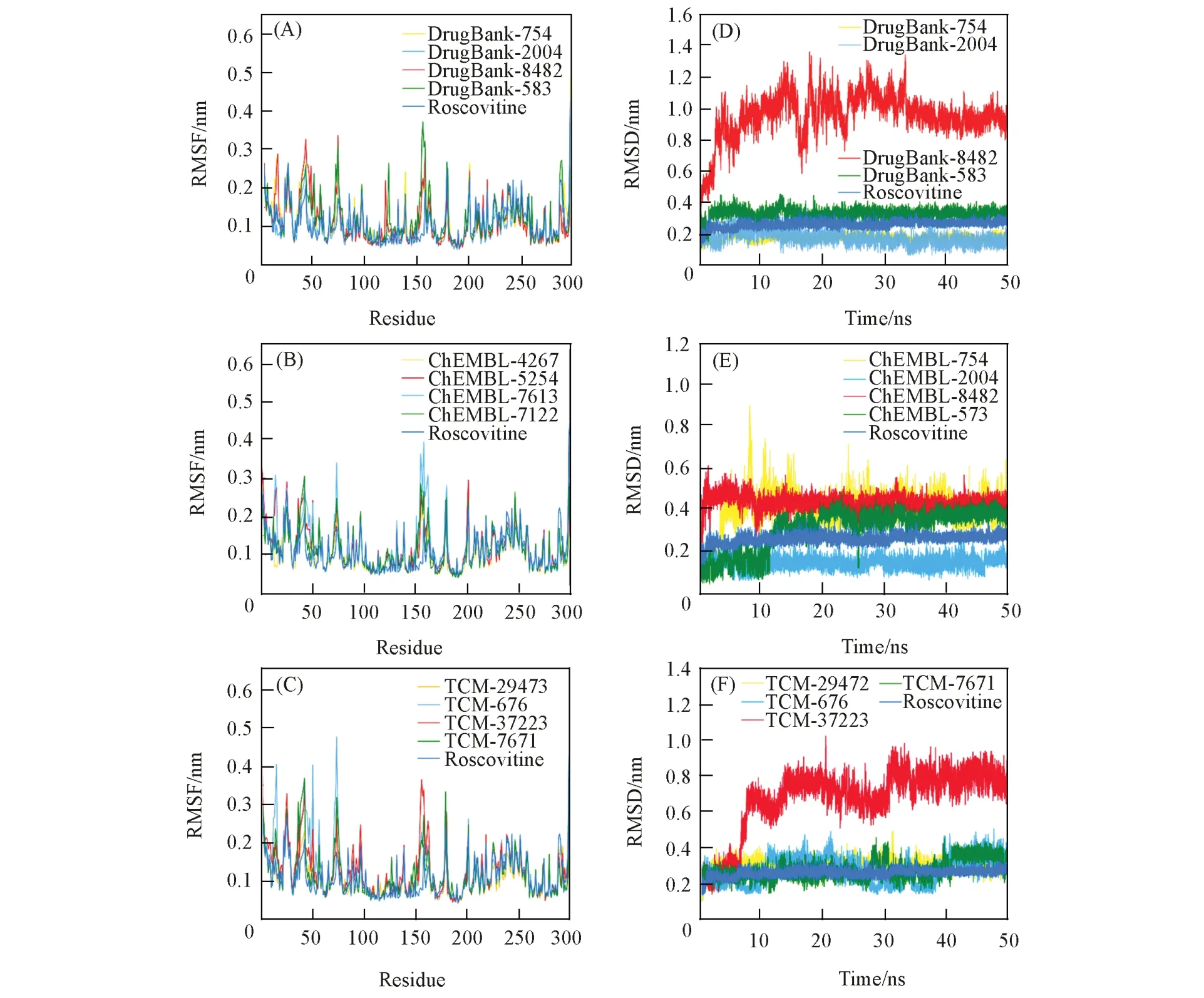
Fig.2 RMSF(A-C)and RMSD(D-F)plots of complexes under MD simulations
通过质心分析(Centroid analysis)研究不同配体与蛋白弱作用的区域.由图S3可见,经过MD模拟后的配体均定位在同一结合位点,说明上述10个苗头化合物与Roscovitine和CDK2/Cyclin A2靶点的结合位点一致.由图S4所示的自由能形貌图可见,这10个苗头化合物的吉布斯自由能最低点的Rg值均在2.02~2.05 nm之间,远小于阳性药物Roscovitine的2.59 nm.说明10个苗头化合物与蛋白质的结合相对紧密.
综合考虑RMSF、RMSD、质心分析和自由能形貌图,发现潜在的10个对CDK2/Cyclin A2复合蛋白有抑制作用的分子,分别为DrugBank-754,DrugBank-2004,DrugBank-583,ChEMBL-4267,ChEMBL-5254,ChEMBL-7613,ChEMBL-7122,TCM-29473,TCM-676和TCM-7671.
2.3 小分子与CDK2/Cyclin A2复合物的结合自由能的分解分析和aNCI分析
进一步采用MM/PBSA方法计算阳性药物Roscovitine以及上述10个候选分子与复合蛋白的结合自由能,发现DrugBank-2004,DrugBank-583和ChEMBL-7122的结合自由能分别为−157.0,−145.3和−150.6 kJ/mol,绝对值明显高于Roscovitine的123.6 kJ/mol,而其余7个分子结合自由能的绝对值均小于Roscovitine.如表2和图S5(本文支持信息)所示.因此在后面分析中只考虑上述这3个分子.
从表2可见,这3个分子与靶点的静电作用分别为−76.7,−115.9和−142.0 kJ/mol,绝对值均大于Roscovitine的57.4 kJ/mol.通过结构分析,发现Roscovitine上的母环是嘌呤基结构,容易与残基形成氢键作用.形成氢键后,芳环的位置被固定,导致环上的取代基与部分残基间产生较大的排斥作用;而DrugBank-2004,DrugBank-583和ChEMBL-7122均含有多个无杂原子的芳环结构,不易与残基形成氢键,构型相对容易改变,因此与CDK2/Cyclin A2之间能产生较强的静电作用.
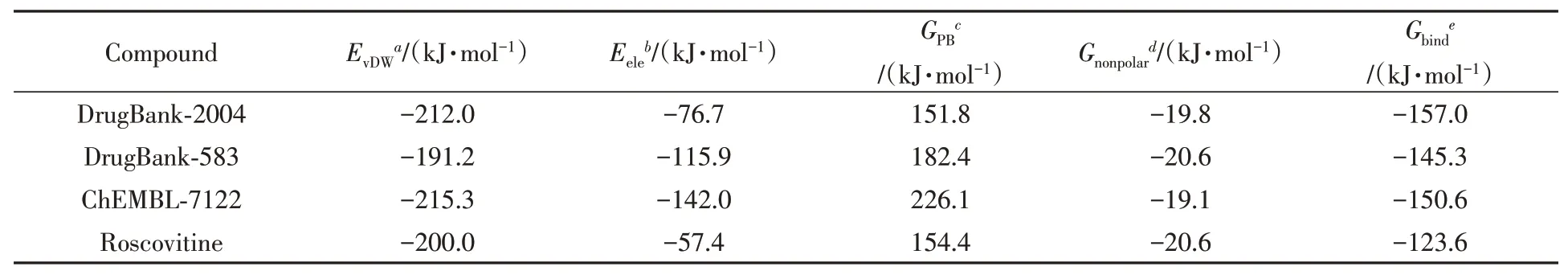
Table 2 Decomposition of binding free energy between hit molecules,the inhibitor Roscovitine and the target protein
截取最后10 ns MD轨迹进行能量分解分析,考察每个残基对总能量的贡献.发现3个先导分子和Roscovitine与复合蛋白具有相似的相互作用.如图3(A)~(H)所示,与Roscovitine类似,Ile10,Glu81,Gln85和Leu134为3个先导分子与靶蛋白作用的主要残基.值得注意的是,这3个先导化合物和Roscovitine与Leu134之间的作用自由能均很大,尤其DrugBank-2004分子,达到−11.0 kJ/mol.综上所述,3个先导分子较阳性药物Roscovitine具有更高的亲和力.
采用MM/PBSA方法进行残基能量分解,对比上述候选化合物及阳性药物Roscovitine与CDK2和CDK2/Cyclin A2蛋白作用的差异,结果如图3和图S5所示.可见,Roscovitine与CDK2的结合自由能较与CDK2/Cyclin A2蛋白的结合自由能大,这是由于CDK2/Cyclin A2结合口袋变大,降低了Roscovitine与残基间的亲合作用.而与阳性药物不同,候选化合物与复合蛋白的结合自由能较与CDK2的结合自由能大,原因是CDK2/Cyclin A2结合口袋变大,削弱了配体与蛋白的Lys33,Glu55,Asp86,Lys129和Asp145残基的排斥作用.
为了直观地显示配体和受体之间的弱相互作用区域,对DrugBank⁃2004,DrugBank⁃583,ChEMBL⁃7122和Roscovitine与CDK2/Cyclin A2形成的复合物进行了aNCI分析.由图4(A)~(D)可见,上述4个分子与CDK2/Cyclin A2蛋白之间等值面的颜色主要是绿色和蓝色,表明配体与CDK2/Cyclin A2复合蛋白的Ile10,Val18和Leu134残基主要通过范德华力和氢键结合.尽管3个候选分子与蛋白的相互作用类型和阳性药物Roscovitine一致,但范德华力和氢键作用明显较Roscovitine强.
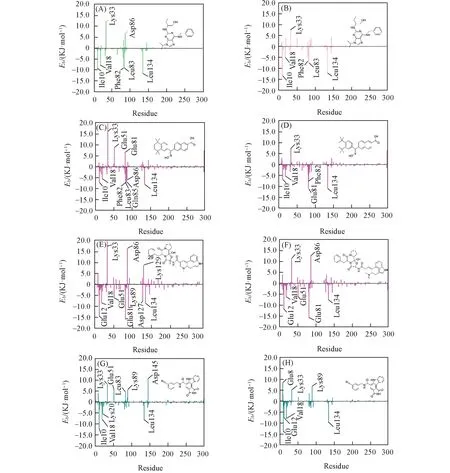
Fig.3 Decomposition of binding free energy on per⁃residue basis into contributions in each systems based on MM/PBSA method
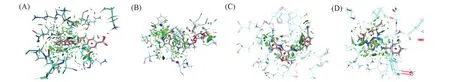
Fig.4 aNCI analysis between CDK2/Cyclin A2 and candidate drug molecules
综上所述,本文通过药效团模型、ADME、分子对接、聚类和毒性预测,从DrugBank,ChEMBL和TCM@Taiwan 3个数据库90万个分子中筛选出12个苗头化合物.基于MD模拟和自由能形貌图结果,发现其中10个分子与CDK2/Cyclin A2复合物能稳定结合.采用MM/PBSA方法发现其中3个先导化合物DrugBank-2004,DrugBank-583和ChEMBL-7122的结合自由能绝对值高于阳性药Roscovitine.利用能量分解和aNCI对自由能贡献进行分析,发现先导分子的结合自由能主要是由范德华力和静电作用构成.由于先导化合物含有多个无杂原子的芳环结构,不易与残基形成氢键,有利于配体的构象转变.因此与靶蛋白的静电作用比Roscovitine强,提高了结合稳定性.通过对比先导分子与CDK2/Cyclin A2和CDK2两种靶点的结合能力,发现它们与CDK2/Cyclin A2形成更稳定的复合物.这是由于CDK2/Cyclin A2的结合位点空间变大,使配体分子与Lys33,Asp86,Lys129和Asp145残基之间的排斥作用减小.本研究的高通量筛选工作能为发现针对CDK2/Cyclin A2靶点的药物研究提供理论依据.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210405.
