基于TCGA数据库分析乳腺癌中TERT相关分子网络机制
2021-10-15陈运景何贵省苏亚静吴灿章敬波吴煌福
陈运景,何贵省,苏亚静,吴灿章,敬波,吴煌福
1.海南医学院研究生院,海南 海口 570100;
2.海南医学院第二附属医院乳甲外科,海南 海口 570100
乳腺癌是全球女性最常见、死亡率最高的恶性肿瘤,据2018 年GLDBOCAN (全球癌症流行病学数据库)估计全球癌症发病率和死亡率显示:女性乳腺癌发病率为11.55% (2 088 849/18 078 957)、仅次于肺癌发病率的11.58% (2093 876/18 078 957)[1-2]。已有研究分析表明,在未来10 年中,中国乳腺癌(breast cancer,BC)的发病率和死亡率将继续上升[3]。随着医疗技术和癌症细胞分子技术发展,可手术治疗的乳腺癌患者在手术切除肿瘤后,通过基因测序、候选突变基因来选择术后的辅助治疗,如化学治疗、雌激素受体调节剂、雌激素受体拮抗剂,免疫抑制剂、靶向治疗、放疗等,乳腺癌患者生存率得到提高。但由于肿瘤原因多样、发病机制尚未明确、生物学侵袭性、病理和分子水平上的异质性,大部分乳腺癌患者因远处转移和相关并发症而死亡,尤其是老年患者[2,4]。众所周知,乳腺癌是一种全身性疾病,以综合治疗为主;乳腺癌的发病机制分子研究已成为国内外研究的主要焦点之一,因此对乳腺癌发生、发展的机制研究,为临床治疗乳腺癌恶性肿瘤的药物研究提供方向,为乳腺癌患者个体化治疗及靶向治疗提供依据。近年来,随着分子生物学和靶向药物的发现,药物的应用,极大地提高了乳腺癌患者的生存时间[5],但部分患者的肿瘤诊断如年龄大、分期高、远处转移、分级高等对预后不良有显著影响[6]。因此,通过分子生物学信息分析了解乳腺癌相关基因表达,探索致病分子机制意义重大。
近年来,许多研究表明,乳腺癌的发生及进展是一个多因素、多阶段、多基因共同参与的变化过程,其中易感基因突变是乳腺癌发生发展重要机制之一[7]。全基因组关联研究(GWAS)已经证明染色体5P15.33上的TERT-CLDTMIL 区域是一个乳腺癌易感基因位点[8],对于乳腺癌相关TERT (telomerase reverse transcriptase 端粒酶逆转录酶)基因的研究已成为当前科研热点。TERT 为端粒酶逆转录酶,是端粒酶中具有生物活性作用的催化亚基,对端粒酶维持端粒长度起重要作用,导致肿瘤细胞无限生长[9]。研究表明,TERT高表达与恶性肿瘤的发生、发展相关,并且与恶性肿瘤的预后不良有关,下调TERT 表达则抑制肿瘤的发展[10]。研究显示,TERT启动子突变导致TERT基因表达上调及基因多态性与前列腺癌预后不良及侵袭性有关[11-12],因此,TERT 基因启动子突变可能成为前列腺癌诊断和预后的生物学标志物。但TERT基因在乳腺癌中的生物学功能及潜在机制研究尚少。
人类癌症基因组图谱(human Cancer Genome Atlas TCGA)数据库是一个广泛的单个癌基因症类型的基因组数据,保存包括基因表达数据、miRNA表达数据、拷贝数变异、DNA甲基化、SNP等数据,为科学研究人员提供了大量、全面的肿瘤基因组和表观遗传学特征数据,识别及综合分析癌症基因组数据和基因组与临床数据的关系及与肿瘤的发生和进展的相关[13-15]。
总之,TERT 表达在恶性肿瘤中起到促癌基因作用;目前TERT基因在乳腺癌发生、发展中的作用研究尚少,具体发病机制未明。本研究通过下载TCGA数据库乳腺癌中miRNA表达数据及临床特征数据,分析TERT在乳腺癌中的作用及致病机制。
1 材料与方法
1.1 数据下载及整理从TCGA数据库下载乳腺癌的miRNA表达数据以及临床资料,共收集1 222份癌标本;其中正常标本113份,乳腺癌肿瘤标本1 109份。将下载的基因表达数据及临床资料数据文件解压成文本文件,利用R软件程序将数据整理为可处理的表达矩阵数据,包括基因名称(Ensembl ID,ensemble 格式名称)和基因表达矩阵文件、临床数据。在ensembl官网(http://asia.ensemble.org/index)中下载人类基因注释文件,在R语言程序中将基因Ensemble ID转化成为Gene ID,以进一步分析。
1.2 TERT 表达差异分析利用R 软件包对下载的mRNA 表达数据进行整合和标准化处理,提取TERT 基因,利用R stats 包Wilcox.test 函数Wilcoxon秩和检验对TERT基因表达量数据在正常和肿瘤分组样本下做差异分析。
1.3 TERT表达与临床病理相关性分析及生存分析将乳腺癌患者中TERT 表达量水平的中位值分为TERT 高表达组与TERT 低表达组,联合临床资料,使用R 软件中survival 包进行生存分析(Kaplan-Meier法),分析乳腺癌中该基因表达量高与患者生存率的关系。为了研究不同的临床特征(年龄、分级、TNM 分期等)和TERT基因表达与乳腺癌患者预后风险的关系,使用coxph 函数进行COX 单、多因素回归分析,并统计P值和HR (风险比)。在COX多因素分析基础上通过R软件中的nomogram函数建立预测生存率的列线图,用Kaplan Meier 生存曲线建立对列线图的预测能力进行验证。
1.4 TERT 共表达分析分析TERT 基因的共表达情况,其中相关性系数过滤条件为0.4,P 值为0.001。 筛选与TERT表达显著的75个基因,用“corrplot”、“circlize”包绘制TERT相关性分析圈图。
1.5 TERT表达与肿瘤微环境及免疫细胞浸润相关性分析使用CIBERSORT 算法[17]对不同亚组的乳腺癌患者RNA-seq数据进行分析,用来推断22种免疫浸润细胞的相对比例,并对基因表达量以及免疫细胞含量进行Spearman 相关性分析。P<0.05 被认为差异有统计学意义。
1.6 TERT表达与药物敏感性分析基于最大的药物基因组学数据库(GDSC 癌症药物敏感性基因组学数据库,https://www.cancerrxgene.org/)[18],使用R 软件包“pRRophetic”来预测每个肿瘤样本的化疗敏感性;用回归的方法得到每种特定化疗药物治疗的IC50估计值,并用GDSC训练集进行10次交叉验证检验回归和预测精度。所有参数都选择了默认值,包括去除批处理效应的“combat”以及取重复基因表达的平均值。
1.7 GSEA 通路富集分析GSEA 分析[19]使用预先定义的基因集,将基因按照在肿瘤样本和正常样本中表达水平的高低进行排序,然后检验该基因集合是否富集在这个排序表的顶端或者底端。本研究通过GSEA比较预先定义的基因集的高风险组及低风险组的信号通路差异情况,探讨两组患者预后差异的可能分子机制,其中置换次数设置为1 000,置换类型设置为phenotype。
1.8 统计学方法TERT 基因在正常样本和肿瘤样本的差异分析使用秩和检验,TERT 表达的乳腺癌患者的生存分析和验证回归模型使用χ2检验和Kaplan Meier 检验;利用Cox 比例风险回归模型进行单因素及多因素分析。所有统计分析均用R 语言(3.6版)进行。所有统计检验均为双侧,以P<0.05 为差异有统计学意义。
2 结果
2.1 TERT基因在正常组织与乳腺癌肿瘤组织中的差异性表达TCGA 乳腺癌数据集分析显示TERT在乳腺癌样本中的表达量高于正常样本,差异有统计学意义(P<0.05),见图1。基于乳腺癌患者性别、肿瘤分期及淋巴结亚组中分析,与正常组织样本相比,TERT 基因在乳腺癌样本中表达显著上调,差异具有统计学意义(P<0.05),见图2。

图1 TCGA 乳腺癌数据集中TERT 在乳腺正常组织和乳腺癌组织中的表达差异

图2 不同性别和肿瘤分期乳腺癌患者的TERT表达水平
2.2 高表达TERT 基因乳腺癌患者的生存率降低基于TCGA数据库及Prognoscan数据库双重验证TERT 基因表达量对乳腺癌患者生存率的影响,以TERT 表达水平中位值为分界点分为高低两组,结果表明TERT高低表达水平两组的生存率差异无统计学意义(P>0.05),见图3A。取TERT 表达水平的最大选择秩统计量分为高低两组,结果表明TERT 表达高低两组的生存率差异具有统计学意义(P<0.05,图3B)。用Prognoscan在线数据库对基因表达综合(GEO)数据集进行验证TERT基因不同表达量对乳腺癌患者的生存影响,其中三个TERT 表达量分组下生存状态差异具有统计学意义,数据集分别为GSE1379 (P<0.05)、GSE11121 (P<0.05)、GSE12276 (P<0.05),见图4。

图3 TCGA数据库TERT基因表达与乳腺癌患者生存率的关系

图4 GEO乳腺癌数据集中TERT基因在乳腺癌组织和正常组织中的差异表达
2.3 临床特征和高表达TERT 对乳腺癌患者预后的影响Cox 单因素、多因素回归分析,如表1 所示,单因素Cox分析结果表明,临床特征如年龄(HR=1.03,P<0.05)、肿瘤分期(HR=2.1,P<0.05)、浸润深度(HR=1.5,P<0.05)、远处转移(HR=6.4,P<0.05)、淋巴结转移(HR=1.7,P<0.05)与乳腺癌患者总生存期相关,而TERT表达(HR=1.2,P<0.05)与总生存期无相关性。多因素Cox分析结果提示年龄(HR=1.03,P<0.05) 和TERT表达量(HR=1.474,P<0.05)与乳腺癌患者总生存期相关。因此,单因素分析提示TERT 表达对患者生存期无相关,但多因素Cox 分析提示有统计学意义,可能TERT 表达对患者生存期影响受到其他因素的影响,表明年龄与TERT 基因为乳腺癌独立预后因素,可能成为重要的预后标志物。在Cox多因素回归分析的基础上,将临床特征如年龄、肿瘤分期、TNM分期和TERT 基因表达量通过nomogram 函数建立一个列线图(图5),对临床特征和TERT 表达量进行评分,将各分值进行相加,得到总分,然后再根据总分来预测TERT 基因表达乳腺癌中的5 年和8 年生存率,对临床治疗和预测乳腺癌患者的预后提供理想模型。为了验证列线图对乳腺癌患者生存率的预测能力,使用Kaplan Meier 曲线建立预测校准曲线图,结果显示如图6 所示,5 年生存率和8 年生存率的预测校准曲线在标准曲线附近,表明列线图的预测能力良好。
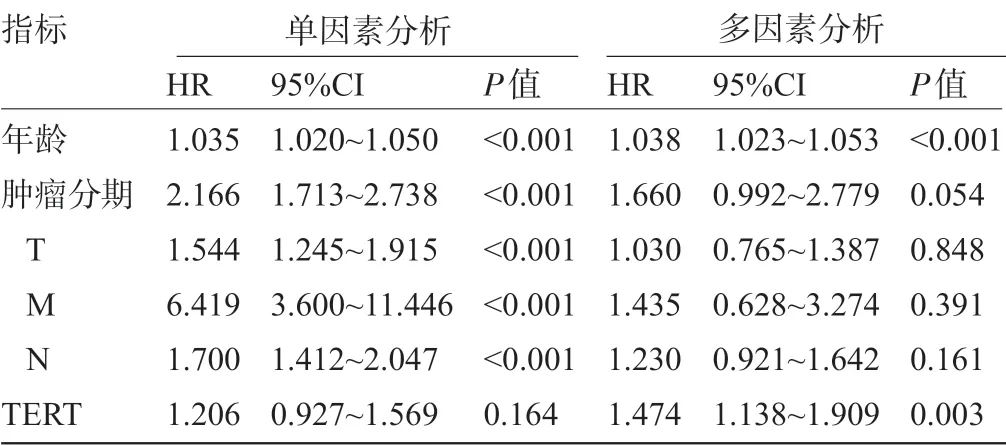
表1 Cox 单因素、多因素分析TERT 表达与临床特征对乳腺癌患者预后的影响

图5 基于临床特征和TERT 基因构建的乳腺癌患者预测5 年和8 年生存率的列线图

图6 标准曲线图
2.4 乳腺癌中TERT 的共表达基因及调控分子进一步根据TCGA 数据库中乳腺癌患者的表达谱,通过相关性分析来探讨乳腺癌中TERT 基因的共表达网络。共筛选出75个与TERT表达显著相关的基因,其中相关性系数正/负相关TOP 5基因热图(图7A)所示,与TERT表达正相关绝对值最大的前5个基因如CNPY1、FSD1、HMGB1P1、ALKAL1、GF1B,与TERT表达负相关绝对值最大的前5 个基因如MCM5、DERA、CACNA1G-AS1、AC004858.1、SLCTA14。 以及共表达相关性圈图如图所示(图7B)。LinkedOmics数据库进一步探讨与TERT潜在调控的miRNA,结果如图所示(图8)。其中正相关前五个miRNA 为hsa-mir-337、hsa-mir-10b、hsa-mir-1245、hsa-mir-199a-1、hsa-mir-377,负相关前五个为:hsa-mir-106b、hsa-mir-550a-2、hsa-mir-92a-2、hsa-mir-1307、hsa-mir-18a。

图7 乳腺癌中TERT基因的共表达基因
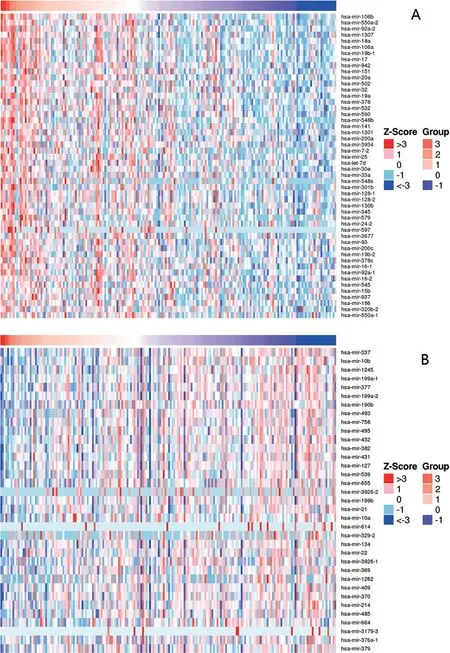
图8 LinkedOmics数据库中TERT潜在调控的miRNA相关性分析
2.5 TERT 表达量与肿瘤微环境及免疫细胞浸润相关性肿瘤微环境主要由肿瘤相关成纤维细胞、免疫细胞、细胞外基质、多种生长因子、炎症因子及特殊的理化特征和癌细胞自身等共同组成,肿瘤微环境显著影响着肿瘤的诊断、生存结局和临床治疗敏感性。通过分析在TCGA 数据集中核心基因与肿瘤免疫浸润的关系,进一步探讨核心基因影响乳腺癌进展的潜在分子机制。使用CIBERSORT 算法和Spearman 相关性分析,分析TERT 与肿瘤微环境的相关性。研究结果表明,TERT与T cells CD4 memory activated、Macrophages M0、Macrophages M1显著正相关,与Mast cells resting、T cells CD4 memory resting 显著负相关(图9A);TERT 与Mast cells resting 的线性关系如图所示(图9B)。

图9 乳腺癌中TERT基因与免疫细胞浸润相关性
2.6 TERT 基因的GSEA 通路富集分析接下来研究TERT 基因涉及的具体信号通路,探讨TERT 影响肿瘤进展的潜在分子机制。GSEA 结果表明,TERT 可富集Mismatch repair、Primary immunodeficiency、RNA polymerase 信号通路(图10),提示TERT可能通过免疫途径影响乳腺癌进展。

图10 乳腺癌TERT基因的Gsea通路富集分析
2.7 乳腺癌患者TERT 表达与药物敏感性分析早期乳腺癌进行手术治疗结合化疗、靶向治疗效果明确。研究基于GDSC 数据库的药物敏感性数据,通过R 包“pRRophetic”来预测肿瘤样本的化疗敏感性,进一步探讨核心基因与常见药物的敏感性。研究结果如图11 所示,TERT 的表达与患者 对Imatinib ( 伊马替尼)、Cisplatin ( 顺铂)、Gefitinib ( 吉非替尼)、Dasatinib ( 达沙替尼)、Erlotinib(埃罗替尼)以及Gemcitabine (吉西他滨)的敏感性相关(P<0.05)。

图11 乳腺癌TERT基因与药物的敏感性分析
3 讨论
乳腺癌是全球女性最常见、死亡率最高的恶性肿瘤,因此研究乳腺癌的发病机制及预后生物分子是必要的,目前研究表明,TERT基因表达与与恶性肿瘤的发生、发展显著相关及预后不良有关[20]。在甲状腺癌中,研究发现TERT基因表达上调与TERT扩增及TERT启动子突变有关,并且与甲状腺不良预后有关[21]。在尿路上皮癌、子宫颈癌、头颈癌中,也研究表明TERT 表达上调与恶性肿瘤的发生、发展相关[22-24]。但TERT基因表达在乳腺癌中研究较少,发病机制尚未清楚。近年来,随着分子诊断技术和科技发展,利用TCGA数据库数据分析乳腺癌中预后的标志物及致病机制,为预测乳腺癌患者预后及研究致病分子机制和治疗提供新思路。
在这项研究中,基于TCGA 数据库[16]中的乳腺癌数据集对TERT基因在乳腺癌中的表达进行了全面和详细的评估,结果表明TERT 基因在肿瘤组织及正常组织中表达的差异,研究发现,TERT基因在乳腺癌样本中表达显著升高,并且TERT高表达乳腺癌患者,其生存期较短。被证实是乳腺癌的独立预后因子[26-27]。
共表达基因是大量功能相关的基因在相关一组条件下有非常相似的表达谱,如被共同的转录因子调控的基因、产物构成同一蛋白复合体、参与相同的调控通路等。因此这些共表达基因在生物学上具有相似的功能。通过基因共表达分析来预测其共表达基因,发现TERT基因表达与CNPY1、FSD1、HMGB1P1、ALKAL1、GF1B 共同促进乳腺癌的发生、发展,其中有研究表明ALKAL1 通过激活SHH 信号通路参与大肠癌的迁徙和侵袭[28]。但FSD1 低表达可促进爱因斯坦- 巴尔病毒(EBV) 相关胃癌的进展[29]。 CNPY1、HMGB1P1、GF1B 等在肿瘤中的生物学功能鲜有研究。分析与TERT基因上调抑制MCM5、DERA、CACNA1G-AS1、AC004858.1、SLCTA14基因的表达参与了乳腺癌的侵袭,其中已有研究发现下调MCM5表达水平通过减少了处于G2期的细胞增殖来抑制乳腺癌的增殖及转移[30]。这与此项研究不一致。需要进一步实验验证。通过LinkedOmics数据库进一步预测可能调控TERT 基因的分子,得出交集的miRNA:hsa-mir-337、hsa-mir-10b、hsa-mir-1245、hsa-mir-199a-1、hsa-mir-377,其中hsa-mir-10b 已经被证实和TERT 启动子突变与神经胶质瘤预后不良有关[31]。通过相关性分析基因共表达基因和潜在调控分子,可以为TERT 可能参与分子机制研究提供实验方向及理论基础。这对于展望目前乳腺癌治疗的进步应该是非常有意义的,可能会发现潜在的生物标志物或预后决定因素的靶点。
肿瘤细胞的组织微环境对肿瘤的发展起着至关重要的作用。肿瘤微环境显著影响着肿瘤的诊断、生存结局和临床治疗敏感性。通过分析在TCGA 数据集中核心基因与肿瘤免疫浸润的关系及核心基因影响乳腺癌进展的潜在分子机制。研究结果表明,TERT与M0 型巨噬细胞、记忆激活的CD4 T 细胞、M1 型巨噬细胞)显著正相关,与静息的肥大细胞、静息的记忆CD4 T 细胞)显著负相关。有研究发现三阴性乳腺癌患者的细胞毒性和记忆性T 细胞显著减少,而调节性T细胞和凋亡T细胞显著增加。肿瘤浸润有极化调节性T细胞提示三阴性乳腺癌患者的免疫抑制微环境和预后较差,认为肿瘤浸润极化调节性T 细胞提示免疫抑制微环境和三阴性乳腺癌患者预后较差[32]。研究发现记忆CD4+T 细胞诱由淋巴结中激活的单核/巨噬细胞产生的,作用于特定肿瘤细胞,从而起到抗肿瘤免疫作用,而且miR-18a 通过抑制CD4+T 细胞的增殖和增加活化诱导的细胞死亡[33-34]。
TERT 基因表达是乳腺癌疾病进展及预后的分子,并且早期乳腺癌手术治疗、化疗、靶向治疗效果明确。因此,进一步探讨核心基因与常见化疗药物的敏感性。研究结果表明,TERT 的高表达乳腺癌患者对伊马替尼、顺铂、吉非替尼、达沙替尼、埃罗替尼以及吉西他滨等药物敏感性高。但低表达hTERT 抑制通过改变白血病肿瘤细胞周期导致肿瘤细胞衰老来增强伊马替尼[35],并且上调hTERT端粒酶活性增加伊马替尼对白血病的耐药性[36]。顺铂在抗肿瘤药物中是属于铂类药物,在肿瘤细胞中可抑制其DNA复制来达到抗肿瘤作用,有较强的广谱抗癌作用。顺铂也可以与其他抗肿瘤药联合用药,如顺铂联合紫衫类药物治疗头颈鳞状细胞癌[37]及间变性甲状腺癌[38]。吉西他宾为胞密啶类抗肿瘤药。研究表明,吉西他宾与hTERT抑制剂联合用药可增加抗肿瘤活性[39]。吉非替尼是一种选择性表皮生长因子受体(EGFR)洛氨酸激酶抑制剂。研究表明吉非替尼通过EGFR 诱导乳腺癌细胞hTERT 下调,使乳腺癌细胞端粒酶活性丧失,使肿瘤细胞失活[40-41]。
综上所述,TERT在乳腺癌中异常上调,其过表达提示这患者预后差及肿瘤的恶性发展,并且主要参与了错配修复、原发性免疫缺陷、RNA 聚合酶信号通路的调控。TERT 基因是乳腺癌治疗的新型靶点,与多种化疗药物、靶向治疗药物敏感性相关。
