艾司洛尔盐酸盐的新合成方法
2021-10-14贺正国江登榜李树生袁明伟
贺正国,蒋 琳,江登榜,岳 越,李树生,袁明伟**
(1. 云南民族大学 化学与环境学院,生物基材料绿色制备技术国家地方联合工程研究中心,云南 昆明 650504;2. 云南南益生物科技有限公司,云南 昆明 650300)
艾司洛尔盐酸盐(Esmolol hydrochloride)是1986 年12 月美国FDA 首次批准生产使用的心血管疾病治疗药物,化学名4-{[3-(1-甲基乙基氨基)-2-羟基]丙氧基}苯丙酸甲酯盐酸盐,其结构如图1所示. 作用机制是通过阻断交感神经系统的β-肾上腺素受体来降低心脏收缩的力度和速度,在临床上主要用作治疗心律失常,降低血压以及缓解心绞痛等[1-2]. 由于艾司洛尔盐酸盐甲酯基团可被红细胞细胞液中的酯酶快速水解,药效半衰期仅10 min,只要停止用药就可以很快消除药效,从而减少或避免可能发生的不良用药反应. 艾司洛尔盐酸盐相较其他常用的β1受体阻滞剂毒副作用小,作用时间短,是一种超短效的β1受体阻滞剂[3-6],艾司洛尔盐酸盐的活性成分是S构型,但是目前成药是以外消旋体的形式入药[7].
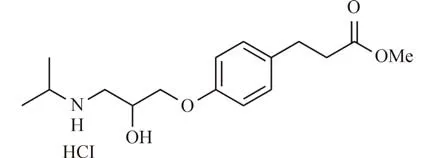
图1 艾司洛尔盐酸盐的化学结构Fig. 1 Chemical structure of esmolol hydrochloride
1982 年Erhardt P W 等以对羟基苯丙酸为原料,首先以硫酸为催化剂合成对羟基苯丙酸甲酯,然后与环氧氯丙烷反应合成化合物3,最后与异丙胺开环反应后和氯化氢成盐合成艾司洛尔盐酸盐,三步总产率14%[6],该方法虽然路线短,但是收率低. 而后便涌现了很多合成方法,如:以对羟基苯甲醛为原料分别与醋酸酐或丙二酸缩合生成对羟基肉桂酸1,经氢化、甲酯化后与环氧氯丙烷及异丙胺反应后,用氯化氢乙醚溶液成盐得到艾司洛尔盐酸盐[8-9],总产率分别为17%、39%(图2,方法A);有文献报道了以对羟基苯甲醛为原料先后与环氧氯丙烷、异丙胺反应后得到中间体4,再和丙二酸缩合后与氯化氢成盐,再甲酯化、氢化得到艾司洛尔盐酸盐,总收率为40%[10](图2,方法B),这两条路线的缺点是产率低,方法B 是先成盐,再甲酯化和氢化,这会导致反应时中间体在溶剂里的溶解度有限,需要大量的溶剂,而且还不利于中间体纯化.
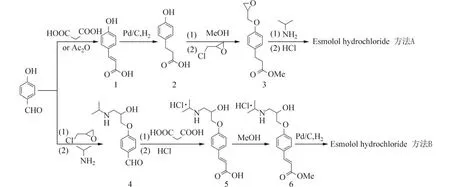
图2 艾司洛尔盐酸盐的合成路线Fig. 2 Synthetic route of esmolol hydrochloride
通过对文献报道艾司洛尔盐酸盐的合成路线进行仔细研究,分析已报道路线的优点和缺点,我们对合成艾司洛尔盐酸盐的路线进行了改进和优化. 选取对溴苯酚为原料,首先是在醋酸钯的催化下与丙烯酸甲酯通过Heck 反应合成对羟基肉桂酸甲酯7,并对此步所使用的碱和溶剂进行了筛选,最优条件下Heck 反应产率为93%,钯碳催化氢化对羟基肉桂酸甲酯得到中间体8,再在碳酸钾的作用下与烯丙基溴醚化得到中间体9,间氯过氧苯甲酸(mCPBA)氧化得到中间体环氧化物3,异丙胺开环氧化物得中间体10,再与氯化氢成盐得到终产品艾司洛尔盐酸盐. 见图3.
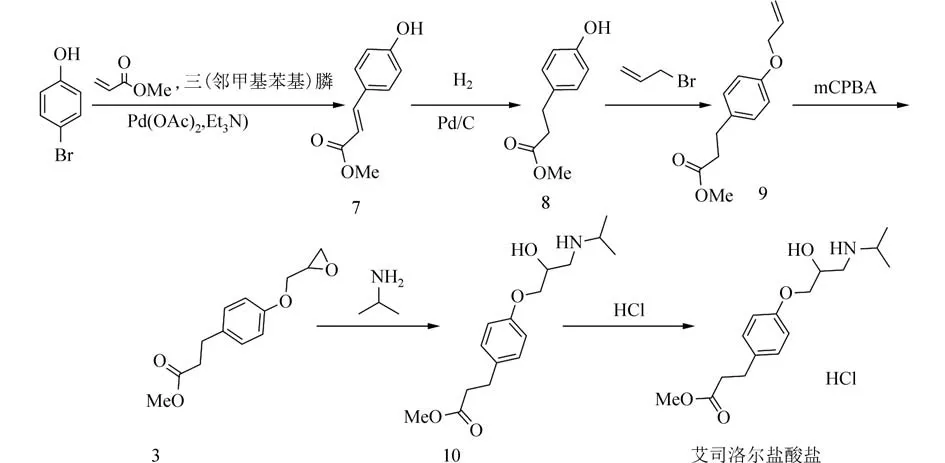
图3 艾司洛尔盐酸盐的合成路线设计Fig. 3 Design of synthetic route of esmolol hydrochloride
1 实验部分
1.1 对羟基肉桂酸甲酯(7)的合成在250 mL 单口瓶中依次加入对溴苯酚(5.00 g,28.90 mmol)、乙腈96.00 mL、三(邻甲基苯基)膦(0.88 g,2.89 mmol)、醋 酸 钯(0.32 g,1.45 mmol)、三 乙 胺(27.10 mL,195.08 mmol),氮气置换反应瓶中的空气,用注射器将丙烯酸甲酯(5.47 g,63.58 mmol)注入到反应瓶中,氮气保护加热回流反应4.0 h. TLC 监控反应,对溴苯酚转化基本完全后停止反应,反应液在旋转蒸发仪上减压浓缩,残余物中加入100.00 mL 乙酸乙酯溶解,加入250.00 mL 水,搅拌10 min,用6.00 mol/L 盐酸调pH 至3.00~4.00,分离有机相,水相用100.00 mL 乙酸乙酯分2 次萃取,合并有机相,饱和氯化钠萃取1 次,无水硫酸钠干燥,减压浓缩,残余物经快速柱层析(乙酸乙酯/石油醚)得白色固体4.77 g,为对羟基肉桂酸甲酯(7),产率93%,熔点:138~140 ℃,化合物7的结构表征数据与文献[11]报道一致.1H NMR(400 MHz,CDCl3)δ:7.64(d,J= 16.0 Hz,1H,PhCHCH),7.42(d,J= 8.6 Hz,2H,Ph-H),6.86(d,J= 8.6 Hz,2H,Ph-H),6.30(d,J= 16.0 Hz,1H,PhCHCH),5.90(s,1H,Ph-OH),3.81(s,3H,―OCH3);13C NMR(101 MHz,CDCl3)δ:168.3,158.1,145.0,130.2,127.3,116.0,115.2,51.9.
1.2 对羟基苯丙酸甲酯(8)的合成在250 mL单口瓶中依次加入对羟基肉桂酸甲酯(4.17 g,23.40 mmol),甲醇78.00 mL,5%钯/碳0.50 g,氢气置换反应瓶中空气,氢气气氛下常温常压搅拌3.0 h,TLC 监控反应,对羟基肉桂酸甲酯反应完毕,抽滤,用少量甲醇淋洗钯碳,滤液在旋转蒸发仪上减压浓缩,得类白色固体4.19 g,为对羟基苯丙酸甲酯(8),产率99%,熔点:39~41 ℃,化合物8 的结构表征数据与文献[12]报道一致.1H NMR(400 MHz,CDCl3)δ:7.04(d,J= 8.6 Hz,2H,Ph-H),6.75(d,J= 8.6 Hz,2H,Ph-H),5.74(s,1H,Ph-OH),3.67(s,3H,OCH3),2.88(t,J= 7.7 Hz,2H,PhCH2CH2),2.61(t,J= 7.7 Hz,2H,PhCH2CH2);13C NMR(101 MHz,CDCl3)δ:174.1,154.4,132.5,129.5,115.5,51.9,36.2,30.2.
1.3 甲基3-[4-(烯丙氧基)苯基]丙酸酯(9)的合成将对羟基苯丙酸甲酯(3.65 g,20.25 mmol),丙酮101.00 mL,碳酸钾(5.35 g,38.69 mmol),碘化钾(0.34 g,2.03 mmol)依次加入到反应瓶中,氮气置换反应瓶内空气,注射器吸取烯丙基溴(5.76 g,47.60 mmol)注入到反应瓶中,室温下反应8.0 h,TLC 监控反应,对羟基苯丙酸甲酯反应完全,抽滤,少量丙酮淋洗滤饼,滤液在旋转蒸发仪上减压浓缩,残余物经快速柱层析(乙酸乙酯/石油醚)得浅黄色油状物3.52 g,为甲基3-[4-(烯丙氧基)苯基]丙酸酯(9),产率79%,化合物9 的结构表征数据与文献[13]报 道 一 致.1H NMR(400 MHz,CDCl3)δ:7.10(d,J= 8.6 Hz,2H,Ph-H),6.84(d,J= 8.6 Hz,2H,Ph-H),6.10~5.99(m,1H,CHCH2),5.43~5.36(m,1H, CHCH2) , 5.30~5.24( m, 1H, CHCH2) ,4.52~4.49(m,2H,OCH2),3.66(s,3H,OCH3),2.89(t,J= 7.8 Hz,2H,PhCH2CH2),2.60(t,J=7.8 Hz,2H,PhCH2CH2);13C NMR(101 MHz,CDCl3)δ: 173.5, 157.3, 133.6, 132.9, 129.3, 117.7, 114.9,69.0,51.7,36.1,30.3.
1.4 4-(环氧乙烷甲氧基)-苯丙酸甲酯(3)的合成将甲基3-[4-(烯丙氧基)苯基]丙酸酯(3.50 g,158.90 mmol),二氯甲烷159.00 mL,间氯过氧苯甲酸(75.0%,4.39 g,19.07 mmol)依次加入到反应瓶中,40 ℃回流反应3.0 h,TLC 监控,甲基3-[4-(烯丙氧基)苯基]丙酸甲酯反应完毕,反应液冷却到室温后加入200.00 mL 30%硫代硫酸钠溶液淬灭反应,搅拌20 min 测溶液不再具有氧化性,静置分层,分出有机相,继续用160.00 mL 二氯甲烷分2次萃取水相,合并有机相用饱和碳酸氢钠萃取1 次,无水硫酸钠干燥、抽滤、浓缩,残余物经硅胶快速柱层析(乙酸乙酯/石油醚)得灰白色固体2.73 g,为4-(环氧乙烷甲氧基)-苯丙酸甲酯(3),产率73%;化合物3 的结构表征数据与文献[14]报道一致.1H NMR(400 MHz,CDCl3)δ:7.11(d,J= 8.7 Hz,2H,Ph-H),6.84(d,J= 8.7 Hz,2H,Ph-H),4.19(dd,J= 11.0,3.2 Hz,1H,Ph-OCH2),3.94(dd,J= 11.0,5.6 Hz,1H,Ph-OCH2),3.66(s,3H,OCH3),3.39~3.28(m,1H,CH2CHOCH2) , 2.94~ 2.83( m, 3H, Ph-CH2,CHOCH2),2.75(dd,J= 4.9,2.7 Hz,1H,CHOCH2),2.59(t,J= 7.8 Hz,2H,Ph-CH2CH2);13C NMR(101 MHz,CDCl3)δ:173.5,157.2,133.4,129.4,114.9,69.0,51.7,50.3,44.9,36.1,30.2.
1.5 4-{[3-(1-甲基乙基氨基)-2-羟基]丙氧基}苯丙酸甲酯(10)的合成将4-(环氧乙烷甲氧基)-苯丙酸甲酯(1.98 g,8.37 mmol)溶解在42.00 mL甲醇中,加入异丙胺(4.95 g,83.68 mmol),加热回流5.0 h,TLC 监控,4-(环氧乙烷甲氧基)-苯丙酸甲酯反应完毕,将反应液在旋转蒸发仪上减压浓缩得微黄色固体2.32 g,熔点:48~50 ℃,为4-{[3-(1-甲基乙基氨基)-2-羟基]丙氧基}苯丙酸甲酯(10),产率94%;化合物10 的结构表征数据与文献[14]报道一致.1H NMR(400 MHz,CDCl3)δ:7.10(d,J= 8.6 Hz,2H,Ph-H),6.84(d,J= 8.6 Hz,2H,Ph-H),4.04~3.97(m,1H,CHOH),3.96~3.93(m,2H,OCH2),3.66(s,3H,OCH3),2.91~2.78(m,4H,CH2CH2),2.71(dd,J= 12.1,7.7 Hz,1H,CH(CH3)2),2.59(t,J= 7.8 Hz,2H,CH2NH),1.08(d,J= 6.3 Hz,6H,CH(CH3)2);13C NMR(101 MHz,CDCl3)δ:173.5,157.4,133.2,129.4,114.8,70.7,68.7,51.7,49.4,49.1,36.1,30.2,23.3,23.1.
1.6 艾 司 洛 尔 盐 酸 盐 的 合 成4-{[3-(1-甲 基 乙基氨基)-2-羟基]丙氧基}苯丙酸甲酯(2.32 g,7.85 mmol)溶解在40.00 mL 甲基叔丁基醚中,氮气置换反应瓶内空气,反应液降温到-5 ℃,缓慢滴加3.00 mol/L 的氯化氢甲醇溶液,滴完后-5 ℃反应1.5 h,体系产生大量的类白色固体,抽滤,少量冷的叔丁基甲基醚淋洗滤饼,五氧化二磷真空干燥得2.38 g 白色固体,熔点:85~86 ℃,为艾司洛尔盐酸盐,产率91%.1H NMR(400 MHz,D2O)δ:7.23(d,J= 8.7 Hz,2H,Ph-H),6.97(d,J= 8.7 Hz,2H,Ph-H),4.37~4.24(m,1H,CHOH),4.19~4.00(m,2H,OCH2),3.64(s,3H,OCH3),3.56~3.41(m,1H,NHCH),3.37~ 3.17(m,2H,CH2NH),2.89(t,J=7.3 Hz,2H,Ph-CH2CH2CO),2.68(t,J= 7.3 Hz,2H,Ph-CH2CH2CO) , 1.36( dd,J= 6.6, 3.1 Hz, 6H,CH(CH3)2);13C NMR(101 MHz,D2O)δ:176.6,156.5,133.8,129.6,114.9,69.7,65.7,52.2,51.9,46.8,35.5,29.4,18.3,17.9.
2 结果与讨论
2.1 对羟基肉桂酸甲酯(7)的合成选择4-溴苯酚和丙烯酸甲酯这两种廉价易得的试剂为初始原料,通过Heck 反应偶联可以高原子经济性地构建目标产物中的丙酸甲酯片段. 催化剂用量为醋酸钯在4-溴苯酚中的摩尔分数x(C4H6O4Pd)计为5%,配体用量为三(邻甲基苯基)膦在4-溴苯酚中的摩尔分数x(C21H21P)计为10%,丙烯酸甲酯用量是4-溴苯酚摩尔数的2.2 倍,在碱存在下反应4.0~5.0 h,经C—C 键偶联生成对羟基肉桂酸甲酯. 对反应中使用的碱及溶剂进行了简单筛选(表1),从表1 中可以看出,在以乙腈作溶剂筛选碱时,三乙胺为碱时产率最高达到了93%,而吡啶作碱时没有反应;接着以三乙胺为碱对溶剂种类进行筛选时,发现只有乙腈作溶剂时反应产率最高,以四氢呋喃作溶剂时,未发生反应,因此4-溴苯酚与丙烯酸甲酯的Heck 反应最佳条件为乙腈作溶剂,三乙胺作碱.
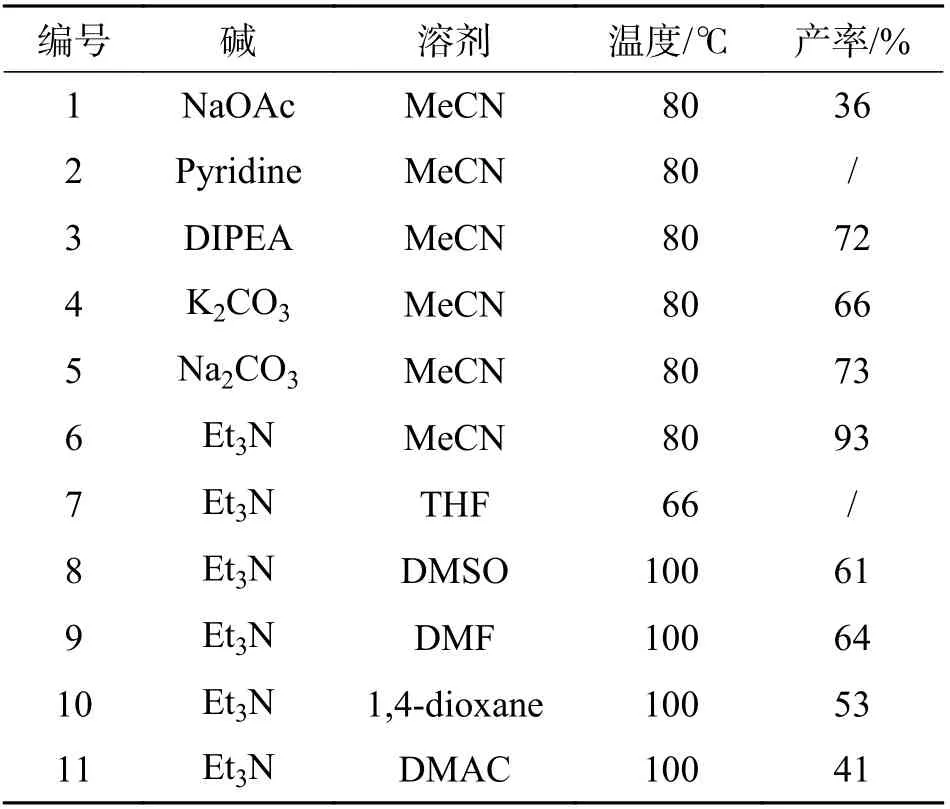
表1 不同的碱及溶剂对Heck 反应的影响Tab. 1 Effects of different alkalis and solvents on Heck reaction
2.2 4-(环氧乙烷甲氧基)-苯丙酸甲酯的合成间氯过氧苯甲酸是富电子烯烃向环氧乙烷转化的常用试剂,特点是反应条件温和,在二氯甲烷等低沸点溶剂中就能将烯烃氧化成环氧. 除了间氯过氧苯甲酸本文还尝试过用双氧水、过氧乙酸等更为廉价易得的氧化剂,但是反应效果不佳,加热回流也无反应发生,所以此步还是间氯过氧苯甲酸较好.
2.3 艾司洛尔盐酸盐的合成艾司洛尔的成盐反应,已报道文献基本采用的是用甲醇作溶剂,然后滴加氯化氢的甲醇溶液,甲醇对艾司洛尔盐酸盐有一定的溶解度,使得产率普遍低于80%. 本文报道的新合成路线采用甲基叔丁基醚作溶剂,-5 ℃滴加3.00 mol/L 氯化氢甲醇溶液直到不再有固体析出为止,产率可达91%.
3 结论
本文以对溴苯酚为起始原料,经过Heck 反应生成对羟基肉桂酸甲酯,氢化后得到对羟基苯丙酸甲酯,再依次与3-溴丙烯醚化,间氯过氧苯甲酸氧化,异丙胺开环得到艾司洛尔游离碱,最后与氯化氢甲醇溶液成盐得到艾司洛尔盐酸盐,总计6 步反应,总收率可达46%. 与传统的方法对比,本路线特点是操作简便,反应条件比较温和,为艾司洛尔盐酸盐的合成提供了一种新思路.
