基于GEO数据库的结肠癌差异基因筛选及其生物信息学分析#
2021-10-12卫静雯成姝婷
卫静雯 成姝婷
(1. 四川大学临床医学院临床医学,四川 成都 610041;2. 四川大学华西基础医学与法医学院,国家卫生健康委员会时间生物学重点实验室(四川大学),四川 成都 610041)
结肠癌(Colorectal cancer,CRC)是一种常见的消化道恶性肿瘤,其在我国的发病率和病死率均居高不下。由于CRC患者确诊时大多数已处于中晚期,因而预后较差,即便是确诊时仍处于早期的患者,其5年生存率也仅为65%;而已发生远处转移的患者,20%~30%在确诊的五年内死亡[1-3]。据我国国家癌症中心分别于2013年、2015年和2019年发布的全国癌症统计数据显示,我国CRC的发病率呈逐年递增的趋势[4]。2018年CRC与直肠癌发病率、死亡率居我国所有恶性肿瘤发病人数的第三位及第五位[5]。癌症的发生与基因转录组的异常存在着密切的关系,通常涉及到多种癌基因、抑癌基因、细胞周期调控基因等的改变,如:相关基因转录组表达水平的变化[6]。本研究通过从美国国立生物技术信息中心 (National Center for Biotechnology Information,NCBI) 公共数据平台Gene Expression Omnibus(GEO) (http://www.ncbi.nlm.nih.gov/geo/)在线数据库获得CRC癌组织和正常癌旁组织的表达谱基因芯片原始数据,分析CRC癌组织和正常癌旁组织之间的差异表达基因(Differentially Expressed Genes,DEGs),以及这些DEGs基因可能参与的生物学功能和信号通路,并初步筛选出与CRC的发生相关的关键基因,旨在探讨CRC的发病机制。
1 资料与方法
1.1 芯片数据获取
在美国国立生物技术信息中心公共数据平台Gene Expression Omnibus (GEO) 数据库(http://www.ncbi.nlm.nih.gov/geo/) 中,以“colon cancer”作为搜索词,检索关于CRC的基因表达数据,数据集纳入条件:CRC患者的转录组RNA表达芯片;芯片组织包含CRC肿瘤组织和与之对应的正常癌旁组织。通过筛选下载了CRC基因芯片数据集GSE44861,该数据集来源于美国国家癌症研究所,贝塞斯达人类致癌实验室,芯片平台为Affymetrix Human Genome U133A Array,包括CRC肿瘤组织和癌旁正常组织的111个结肠组织的基因表达谱。
1.2 方法
1.2.1 数据预处理
通过active perl软件对原始数据进行处理,包括单个样本数据汇总为矩阵、探针ID转换为Gene symbol。
1.2.2 差异基因筛选
使用R软件Limma包(Linear Models for Microarray Data)对预处理后的芯片数据进行分析,设置假阳性概率(False Discovery Rate,FDR)<0.05和样本件差异倍数 |logFC| >1.0为筛选阈值,筛选CRC肿瘤组织和癌旁正常组织间的差异表达的DEGs。
1.2.3 基因本和信号通路富集分析
使用Database for Annotation, Visualization and Integrated Discovery(DAVID)数据库(http://david.abcc.ncifcrf.gov/)的在线分析工具对筛选出的DEGs进行富集分析,包括基因本(Gene Ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)信号通路[7,8]。其中GO分析对生物过程(Biological Process,BP)、分子功能(Molecular Function,MF)和细胞组成(Cellular Component,CC)进行了富集。设定FDR<0.001为筛选阈值。
1.2.4 蛋白互作网络构建
为研究DEGs所编码蛋白质之间的相互关系, 本研究使用了STRING数据库 (http://www.stringdb.org/) 对筛选出的DEGs编码蛋白进行了蛋白互作(Protein-protein interaction,PPI)分析,设定置信度 > 0.7,并构建PPI网络。采用Cytoscape软件(v3.8.0)的Cytohubba插件对PPI结果进行可视化分析,以MCC算法获取前10位关键基因[9]。
1.3 统计学方法
本研究中基因芯片表达数据采用R软件(v4.0.2)进行差异基因分析,采用DAVID在线工具对差异基因进行基因本和信号通路富集分析,采用STRING在线工具对差异基因编码蛋白进行PPI分析,采用Cytoscape软件(v3.8.0)的Cytohubba插件对PPI结果进行关键基因分析。P<0.05表示差异有统计学意义。
2 结果
2.1 CRC组织DEGs筛选结果
分析GSE44861基因表达谱,共筛选出241个DEGs,其中74个基因表达上调,167个基因表达下调。见图1。

图1 GSE44861基因表达谱中差异基因。A为差异基因的火山图,B为前50个差异基因的热图。
2.2 DEGs的GO和KEGG富集分析结果
GO富集分析结果表明,DEGs在8条BP中显著富集:胃肠上皮的维护(Maintenance of gastrointestinal epithelium)、体液免疫反应(Humoral immune response)、细胞外结构组织(Extracellular structure organization)、细胞外基质组织(Extracellular matrix organization)、消化(Digestion)、碳酸氢盐运输(Bicarbonate transport)、抗菌体液反应(Antimicrobial humoral response)、抗菌肽介导的抗菌体液免疫应答( Antimicrobial humoral immune response mediated by antimicrobial peptide),见图2a;差异基因在5条cc中显著富集:微绒毛膜(Microvillus membrane)、微绒毛(Micronillus)、含胶原细胞外基质(Collagen-containing extracellular matrix)、顶端质膜(Apical plasma membrane)、细胞顶端(Apical part of cell),见图2B;差异基因在13条MF中显著富集:信号受体激活剂活性(Signaling receptor activator activity)、受体配体活性(Receptor ligand activity)、氧化还原酶活性,作用于供体的CH-OH基团,NAD或NADP为受体(Oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as accetor)、氧化还原酶活性,作用于供体的CH-OH基团(Oxidoreductase activity, acting on the CH-OH group of donors)、激素活性(Hormone activity)、糖胺聚糖结合(Glycosaminoglycan biding)、具有抗张强度的细胞外基质结构成分(Extracellular matrix structural constituent conferring tensile strength)、CXCR趋化因子受体结合(CXCR chemokine receptor binding)、趋化因子受体结合(Chemokine receptor binding)、趋化因子活性(Chemokine activity)、碳酸盐脱水酶活性(Carbonate dehydratase activity)、阴离子跨膜转运蛋白活性(Anion transmembrane transporter activity),见图2C。此外, 差异表达基因在2条KEGG 通路中富集: 氮代谢(Nitrogen metabolism)、胆汁分泌(Bile secretion),见图2D。

图2 差异基因的功能和通路富集分析。A表示BP;B表示CC;C表示MF;D表示KEGG。
2.3 差异基因的PPI网络
将所有DEGs对应蛋白列表上传STRING数据库分析其相互作用关系,PPI结果显示,该PPI网络共有238个节点,208条边线。通过Cytoscape软件得到该PPI网络中的10关键蛋白分别为:AGT、CXCL1、CXCL2、CXCL3、CXCL11、CXCL13、SST、PYY、P2RY14、INSL5,见图3。
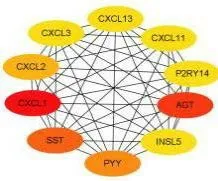
图3 差异基因表达蛋白PPI网络的关键蛋白
2.4 差异基因的PPI网络
将所有DEGs对应蛋白列表上传STRING数据库分析其相互作用关系,PPI结果显示,该PPI网络共有238个节点,208条边线。通过Cytoscape软件得到该PPI网络中的10关键蛋白分别为:AGT、CXCL1、CXCL2、CXCL3、CXCL11、CXCL13、SST、PYY、P2RY14、INSL5,见图3。
2.5 关键基因(Hub gene)在DEGs富集结果中的分布
在DEGs富集结果中各GO注释和KEGG通路包含的DEGs中查找由PPI网络获得的10关键蛋白所对应的hub基因,发现除了P2RY14外,其余9个hub基因主要分布于3条BP(抗菌肽介导的抗菌体液免疫应答、抗菌体液反应、体液免疫反应)和6条MF(受体配体活性、信号受体激活剂活性、CXCR趋化因子受体结合、激素活性、趋化因子受体结合、趋化因子活性)。所有的Hub基因在CC和KEGG富集结果中没有分布。
3 讨论
CRC的发生、发展与相关基因转录组的改变有着密切的关系[6]。本研究利用GEO数据库中CRC组织和相应癌旁组织的基因芯片数据对其DEGs进行了筛选,共获得241个DEGs,其中74个上调,167个下调。我们首先利用DAVID数据库分析了DEGs在BP、CC、MF和KEGG中的富集情况,得到8条BP、5条CC、13条MF和2条KEGG注释;之后利用STRING数据库对这些DEGs对应蛋白质进行了分析,得到了它们之间的相互作用关系,并采用Cytoscape软件的Cytohubba插件筛选出了10个关键蛋白的hub基因。在这些hub基因中,P2RY14未能在GO和KEGG中富集,可能与CRC中改变的BP、CC、MF和KEGG关系不大;但其表达水平在CRC与癌旁组织之间的差异提示该基因本身与CRC存在一定的联系,只是这种联系的发生与BP、CC、MF或信号通路无关。现有研究虽尚无P2RY14与CRC的报道,但其与其它肿瘤有着紧密的联系,如:P2RX7等嘌呤能受体可增加肿瘤的侵袭和转移,是不良预后因素[10,11]。
本研究还发现,富集于“CXCR趋化因子受体结合”MF注释的所有DEGs均是hub基因;而富集于“趋化因子受体结合”和“趋化因子活性”的MF注释大部分DEGs为hub基因,提示CRC的发生与趋化因子受体结合(特别是CXCR趋化因子受体结合)有着紧密的关系。趋化因子及其受体家族是白细胞迁移至炎症部位的重要介质,长期炎症可能为肿瘤细胞的发育和生长提供理想的微环境,趋化因子家族可以通过吸引肿瘤促进和抗肿瘤细胞来刺激或抑制肿瘤的发展[12]。有研究表明,CXC趋化因子家族被白介素-1α在结肠癌细胞系中强烈诱导,表明了结肠癌与炎症过程的潜在联系,并在肿瘤生长调节与转移中有重要作用[13]。CXCL1、CXCL2 、CXCL3、CXCL11、CXCL13均属于CXC趋化因子家族的小细胞因子,且在不同肿瘤的发生发展有着重要的作用。目前已知CXCL3在包括CRC在内的多种侵袭性肿瘤中过表达,是肿瘤发生发展的重要因子[14];CXCL1也可能与结肠癌的发生相关[15];CXCL11在CRC中的作用存在争议,有研究者认为在CRC细胞系中过表达,可抑制细胞毒T淋巴细胞向肿瘤细胞的迁移,与CRC的迁移、侵袭、转移和体外上皮间质转化相关,可能是结肠癌患者总生存期较差的危险因素[16-18];另一些研究者认为CXCL11可促进抗肿瘤免疫从而提高生存率,可作为结肠腺癌患者的独立良性预后生物标志物[16]。CXCL2和CXCL13与炎症和免疫有着密切的联系,但尚未确定与CRC的关系[20,21]。
此外,本研究中hub基因还多富集于“抗菌肽介导的抗菌体液免疫应答”、“抗菌体液反应”、“体液免疫反应”的BP注释和“受体配体活性”、“信号受体激活剂活性”和“激素活性”的MF注释,提示CRC的发生或与肠道感染和免疫应答有关。
综上所述, CRC的发生可能与CXCR趋化因子受体结合以及肠道感染和免疫应答密切相关,P2RY14作为CRC中较为独立的DEGs,也极有可能成为CRC的筛查、诊断、预后的分子生物标志物。但本研究仅利用生物信息学分析进行初步筛选,且测序数据的样本量较小,存在一定的局限性,后期需进一步实验验证。
