锶内标法在溶液法制样-X射线荧光光谱法测定天然铀产品(U3O8)中铀的可行性研究
2021-10-11柳金良宋茂生岳中慧范鹏飞
柳金良,宋茂生,张 鑫,岳中慧,范鹏飞
(核工业二三〇研究所,长沙 410007)
在天然铀产品(U3O8)贸易中对铀进行检测时,质控样品中铀的绝对误差需控制在0.1%以下,才能认为铀的测定结果是准确有效的。因此,建立准确测定天然铀产品(U3O8)中铀含量的方法至关重要。
测定天然铀产品(U3O8)中铀含量的方法主要有滴定法[1-3]、溶液法制样-X 射线荧光光谱法(XRFS)。采用滴定法时,在磷酸和硫酸混合溶液介质中先用亚铁盐将U6+还原成U4+,再以钼为催化剂用硝酸将多余的亚铁盐氧化,过程中产生的氮氧化物用氨基磺酸消除,然后加入硫酸钒酰溶液,以二苯胺磺酸钠作指示剂,用重铬酸钾溶液滴定法测定铀含量。滴定法操作繁琐,耗时较长,且测定值受环境和人为因素影响较大。溶液法制样-XRFS 是先将样品消解为酸性溶液,然后采用XRFS测定其中目标物含量的方法,该方法可以消除样品间密度和成分的不均匀性,以及粒度、矿物效应,同时可降低各组分间的吸收-增强效应[4],常用于分析高铀含量的样品[5-10],铀测定值的相对标准偏差(RSD)一般为0.32%~5.0%。但用这种方法分析标准物质GBW 04205 时,测定值与认定值的绝对误差大于0.27%,无法满足天然铀产品(U3O8)贸易关于质控样品的检测要求。
在用XRFS测定时,内标法具有精确度高的优势。文献[11-12]用XRFS-内标法同时测定钨精矿中WO3和铁矿石中铁元素,XRFS的测定值和化学法的结果基本一致,RSD 分别为0.069%,0.11%;文献[13]研究了内标法在XRFS测定土壤重金属镍元素中的应用,发现内标法可有效提高测定值的准确度。
因此,本工作以溶液法制样-XRFS测定高含量铀的文献[5-10]为参考,引入与铀具有相似谱线的锶作为内标,建立了溶液法制样-XRFS测定天然铀产品(U3O8)中铀含量的方法,并分析了锶内标法的可行性,以期为天然铀产品(U3O8)的验证方对铀含量的准确测定提供参考。
1 试验部分
1.1 仪器与试剂
AxiosMAX型波长色散X 射线荧光光谱仪,配最大管功率为4.0 kW,最大管激发电压为60 kV,最大管电流为160 mA,SSTMAX 超尖锐端窗型铑靶X 光 管,附氦气光路系统和Super Q5.1A 软件;BT125D 型电子天平(精度0.01 mg),应在恒温恒湿、封闭的环境中称量,以避免震动和气流波动的影响;CJJ78-1型磁力搅拌器;YP3102型电子秤(精度0.01 g);塑料样杯(用于装载溶液),内径37 mm;Mylar®麦拉膜(一种高分子聚酯薄膜),厚度6μm;钢杯(用于装载塑料样杯,可被液体传感器感应)材质为不锈钢,内径40 mm。
U3O8成分分析标准物质GBW 04205,其中铀认定值84.711%,标准不确定度为0.021%。
氦气纯度大于99.99%;碳酸锶的纯度大于99.99%;硝酸、磷酸均为分析纯;硫酸锶为优级纯;试验用水为二次去离子水。
1.2 仪器工作条件
由于本方法属于高精度测定方法,应优先选择色散度高的分光晶体LiF220;双背景模式需保证有足够的测量时间。选用高性能闪烁探测器Hiper-Scint,最大探测计数率为3 750 kcps,选用的滤光片材质为铝,厚度为200μm。其他XRFS工作参数见表1,其中PHD 为脉冲高度分布电压。
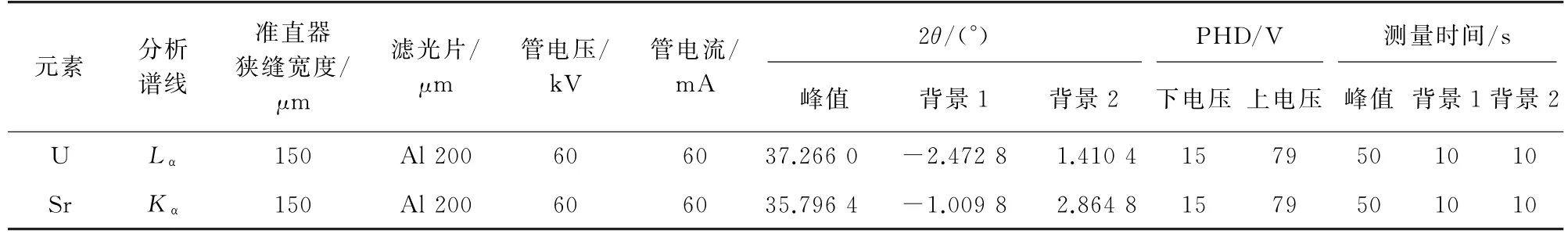
表1 XRFS工作参数Tab.1 Working parameters of XRFS
1.3 试验方法
将碳酸锶于105℃干燥4 h,称取0.600 00 g碳酸锶和1.000 00 g天然铀样品于250 mL 锥形瓶中,用少量水冲洗杯壁,加入15 mL磷酸和2 mL硝酸,在250 ℃电炉上加热约20 min,取下稍冷,加水至100 mL,再加入10 mL硝酸,在250 ℃电热炉上加热10 min。取下冷却,放入一粒磁子,用水冲洗杯壁并加水至200 mL,磁力搅拌1 min,用一次性塑料吸管吸取20 g(精确至0.01 g)溶液于塑料样杯中,按照仪器工作条件测定。随同制备空白溶液。
2 结果与讨论
2.1 锶作内标的可行性分析
2.1.1 天然铀产品(U3O8)中的锶含量的影响
样品中不能含有内标元素是选择内标元素的原则之一[14]。U3O8粉末是经铀矿石加工提纯后的天然铀产品,在生产中,已对其中的杂质进行了有效控制[15],大部分杂质元素含量处于微量或痕量水平[15-20],但都不涉及锶元素,而天然铀产品(U3O8)贸易所检测的杂质指标中也不包含锶元素。为探究样品中锶的含量水平,试验从不同批次样品中抽取20个具有代表性的样品,按照文献[16]使用电感耦合等离子体质谱法(ICP-MS)测定其中的锶含量,结果见图1。

图1 天然铀样品中锶含量Fig.1 Strontium content in natural uranium sample
由图1 可知,样品中锶的质量分数不大于8.0μg·g-1,与硝酸锶称量值(0.600 00 g)相比,样品中的锶含量对测定值的影响可以忽略。
2.1.2 锶内标分析谱线的选择
为确保内标元素和分析元素的吸收-增强效应基本相同,需满足以下条件:分析元素谱线和内标元素谱线及其吸收限的相对关系应保持一致,互不发生干扰;内标元素和分析元素的特征X 射线波长均位于基体元素特征X 射线波长及其吸收限的同一侧;分析元素和内标元素的吸收限波长接近[21]。据此,本工作采用的铀的分析谱线为Lα,选择相邻谱线锶元素的Kα为内标特征线,其中铀的Lα波长为0.091 1 nm,吸收限为0.072 2 nm;锶的Kα波长为0.087 7 nm,吸收限为0.077 0 nm。分析元素、内标元素以及基体元素谱线与其对应的吸收限位置见图2。

图2 锶、铀以及基体谱线与吸收限的位置Fig.2 Position of the spectral lines and the absorption limits of Sr,U and matrix
由图2可知,铀和锶的分析谱线和吸收限均比较接近,且位于轻基体(样品溶液中的主要基体组分为水、磷酸、硝酸,及杂质元素钠、硅、铁、钙、硫等,均属于轻基体)特征X 射线和吸收限的同一侧,说明锶和铀的吸收-增强效应基本一致。因此,可采用锶元素的Kα作为内标特征线。
2.1.3 不同浓度下锶、铀的荧光强度的变化趋势
分析谱线的波长接近时,分析元素和内标元素的谱线强度应在相同浓度水平下保持一致,否则不能用作内标[14]。用5%(体积分数)硝酸溶液稀释1.0 mol·L-1锶和铀的标准溶液,配制锶和铀的混合标准溶液系列,浓度分别为0.002,0.004,0.01,0.02,0.03,0.04,0.06 mol·L-1,按照仪器工作条件测定,结果见图3。其中I为荧光强度。
由图3可知,铀、锶的荧光强度随浓度的增加而增加,但浓度和荧光强度的关系曲线均向下弯曲,可能与元素的吸收-增强效应有关;在0.002~0.06 mol·L-1内,铀、锶的荧光强度的比值为1.1~1.2,与浓度基本呈直线分布,说明在浓度相同的情况下,铀和锶谱线强度基本相同。因此,锶可以作为测定铀的内标元素。
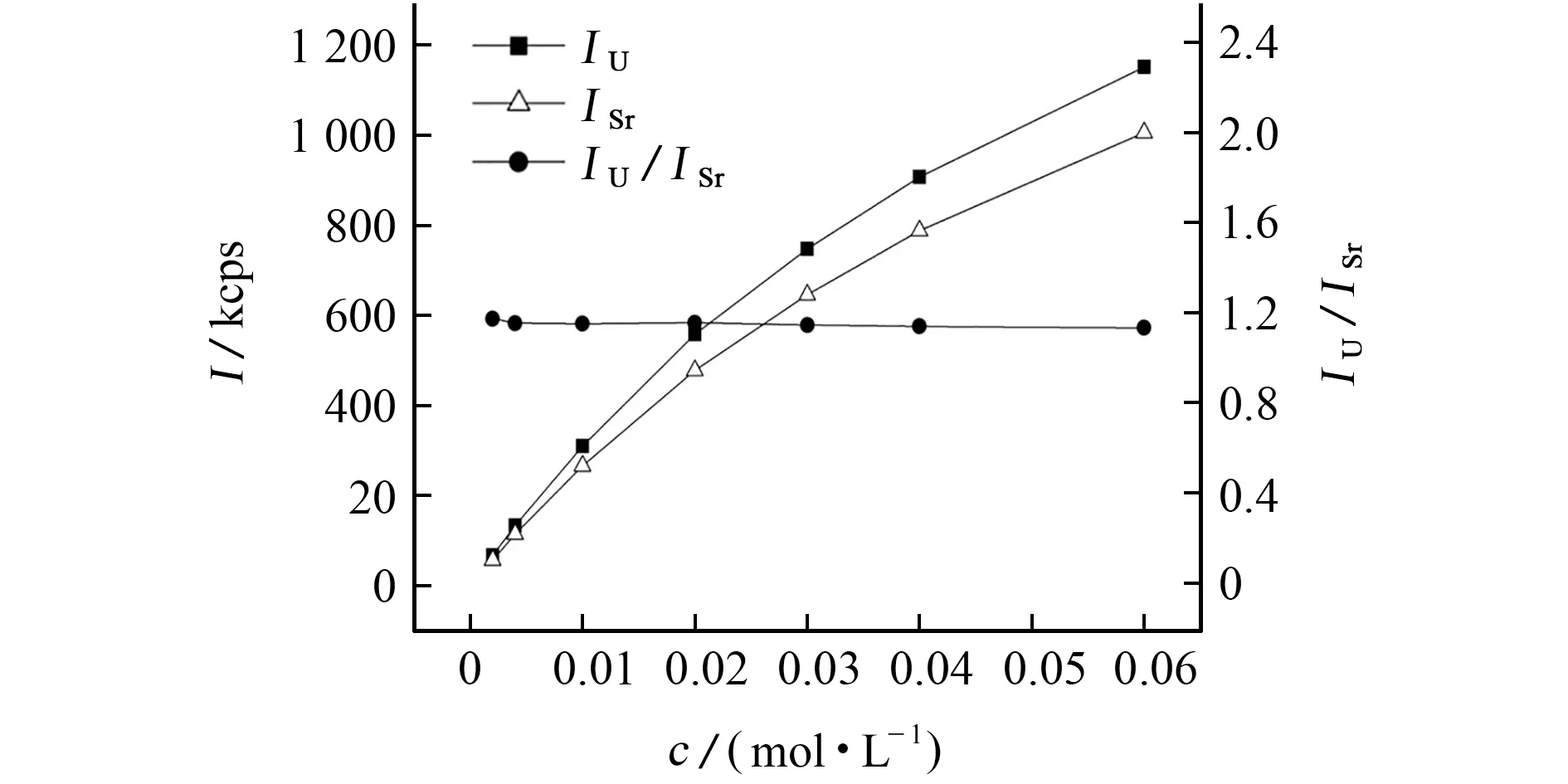
图3 不同浓度下锶、铀测定值的变化Fig.3 Changes of the determined values of strontium and uranium at different concentrations
2.1.4 锶内标化合物的选择
内标物应是一种已知含量的标准物质或高纯物质,并能和样品较好地混合且容易获得。实验室常见的含锶化合物有硫酸锶、碳酸锶、硝酸锶和氯化锶等。硝酸锶微溶于水,在酸性溶液中易沉淀;氯化锶在空气中易形成带结晶水的化合物;硝酸锶是一种易制爆化学品,且高纯级别的较难获得。因此,试验选择纯度大于99.99%且易溶于酸性溶液的碳酸锶作为锶内标化合物。
2.1.5 硫对锶内标的影响
天然铀产品(U3O8)中含硫酸根的化合物会与锶内标化合物反应形成硫酸锶沉淀[22-23],进而影响铀的测定结果。为了考察硫的影响,在样品溶液中滴加硫酸,当滴加到一定量时,溶液中会产生白色沉淀,说明硫酸锶沉淀的形成和样品中硫酸根的含量有关,故应明确样品中硫的最大允许含量。依据文献[24]测定固体溶解度的方法,分别加入0.500 00 g硫酸锶固体,按照试验方法制备6 份样品溶液,用定量滤纸过滤,称取未溶解的硫酸锶的质量,计算所得硫酸锶的溶解量为0.348 12 g,从而计算出样品含硫酸根的化合物中的最大允许含硫量为6.06%,而历年(2015-2019)实验室分析的天然铀产品(U3O8)中总硫含量的水平见图4。
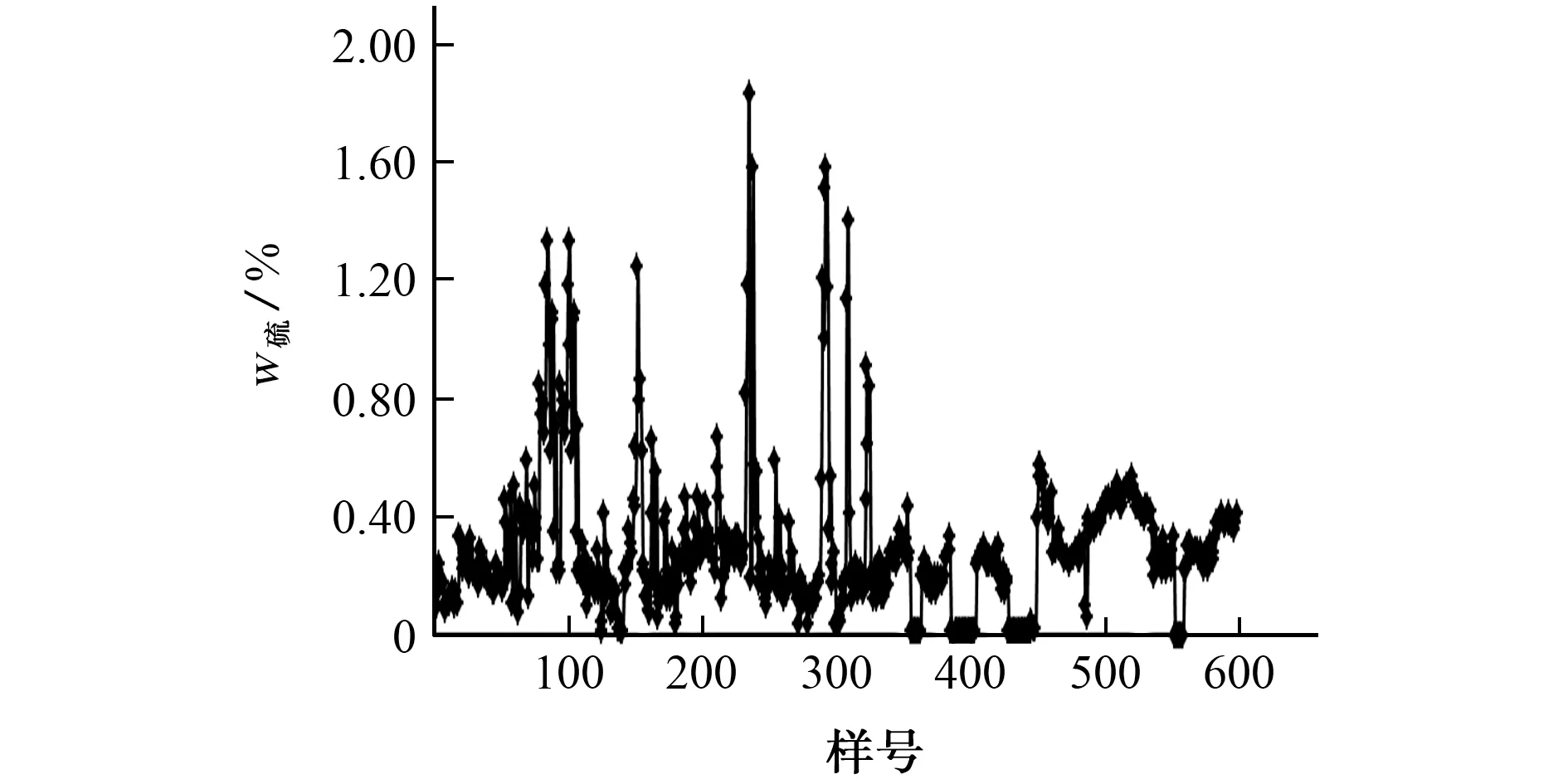
图4 实验室分析的天然铀产品(U3 O8)中总硫含量水平Fig.4 Total S level of natural uranium samples(U3 O8)analyzed by laboratory
由图4 可知,样品中总硫的质量分数大多在0.50%以下,最高为1.83%,均远低于6.06%,而含硫酸根的化合物中的硫低于样品中的总硫含量,说明日常样品中硫的含量水平不会对锶内标法的测定产生干扰,但是如果增大样品的称样量(大于1.000 0 g)或者降低样品溶液的体积(小于200 mL)时应重新考虑硫的潜在影响。
2.2 定容体积、进样量变化和仪器波动对锶内标法测定的影响
在采用标准曲线法定量时,样品溶液定容体积、进样量及仪器波动对测量结果影响较大,而内标法可降低其影响。综合考虑测定过程中可能涉及的定容体积、进样量及仪器波动的变化幅度,设计以下3个试验:取20 mL 样品溶液6份,分别加0,1,2,3,4,5 mL 水对其进行稀释,取稀释后的溶液20 g上机测试,计算IU/ISr和其对应的RSD,以考察定容体积的变化对测定的影响;取17,18,19,20,21,22 g样品溶液直接上机测试,计算IU/ISr和其对应的RSD,以考察进样量的变化对测定的影响;取20 g样品溶液1份,直接上机重复测定6次,计算IU/ISr和其对应的RSD,以考察仪器波动对测定的影响。结果显示:3种试验条件下,IU/ISr分别为1.018 5,1.018 0,1.017 6,结果比较接近,且都趋近于1;RSD 分别为0.039%,0.032%,0.033%,均小于0.040%,且结果比较接近,说明采用锶内标法可以消除常规试验中定容体积、进样量和仪器波动的变化对测定结果的影响,同时也说明了在样品消解时以锥形瓶定容的方法可行,避免了转移定容带来的误差和污染,简化操作流程,适合批量化检测。
2.3 标准曲线
将标准物质GBW 04205在850 ℃下灼烧4 h,以一定的浓度梯度称取 GBW 04205 和约0.600 00 g(理论称样量)碳酸锶,实际称样量见表2,以样品理论称样量(1.000 00 g)进行换算,对应的铀的质量分数为78.78%~87.25%,符合实验室分析的天然铀产品(U3O8)中铀的含量范围。按照试验方法配制标准溶液系列,按照仪器工作条件测量铀和锶的荧光强度。以IU/(ISr×0.600 00/mSr)为纵坐标,84.711%(GBW 04205 中铀的认定值)与标准物质的实际称样量的乘积为横坐标绘制标准曲线。结果显示:铀的线性范围为0.788 57~0.873 62 mg,线性回归方程为y=0.011 93x+0.004 870,相关系数为0.999 8。标准曲线参数见表2。
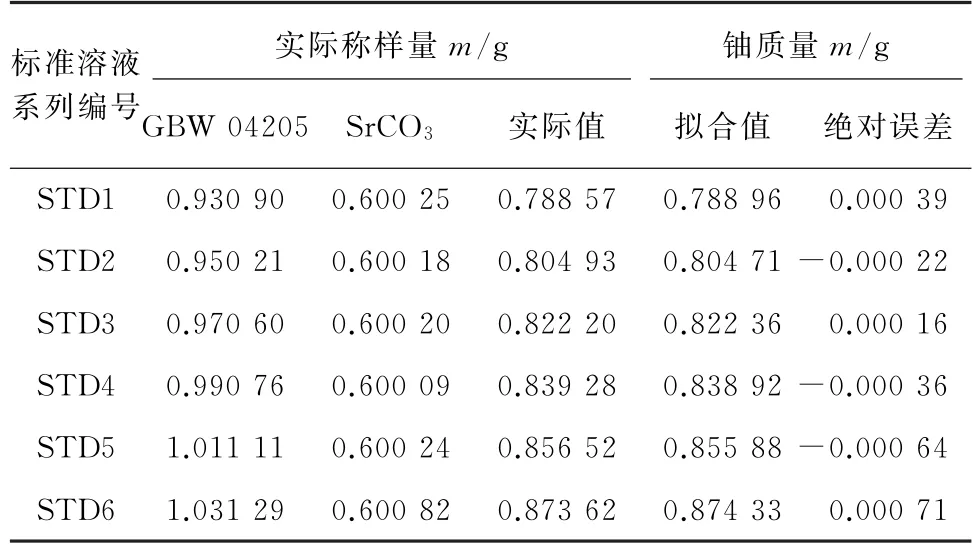
表2 标准曲线参数Tab.2 Parameters of calibration curve
由表2可知,铀质量的拟合值和实际值的绝对误差小于0.001 g,结合标准曲线的相关系数,说明以锶内标法建立的线性关系较好。
2.4 基体效应
由于消解后的样品溶液中含有酸消解带来的杂质及其他杂质,需要进行基体校正,校正公式见式(1):
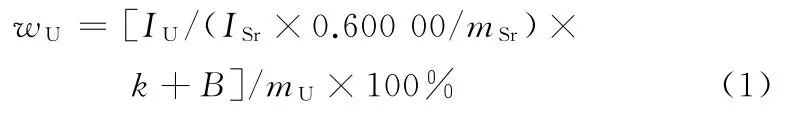
式中:wU为铀元素质量分数;IU为铀元素的荧光强度;ISr为锶元素的荧光强度;k为标准曲线的斜率;B为标准曲线的截距;0.600 00 g为内标物碳酸锶的理论称样量;mSr为内标物碳酸锶的实际称样量;mU为样品的实际称样量。
2.5 精密度和准确度试验
将标准物质GBW 04205在850 ℃下灼烧4 h,称取约1.000 00 g GBW 04205,按照试验方法制备11份样品溶液并测定,将测定值带入公式(1)以消除基体效应,计算校正后的测定值的RSD,并与认定值进行比较。结果显示:校正后的铀的质量分数为84.722%,与认定值的绝对误差为0.011%,小于天然铀产品(U3O8)贸易检测时的质控要求(0.1%),RSD 为0.061%,说明以锶内标法建立的方法的精密度和准确度较好。
2.6 样品分析
本方法用于6个杂质元素含量变化较宽的天然铀产品(U3O8)的分析,并与国家标准GB/T 11848.1-1989«重铀酸盐中铀的测定 硫酸亚铁还原/重铬酸钾氧化滴定法»中的电位滴定法[2]进行对比。结果显示:样品1~样品6 的测定值分别为82.369%,79.462%,82.954%,80.493%,83.161%,84.575%,电位滴定法测定值分别为82.391%,79.501%,82.891%,80.637%,83.069%,84.684%,两种方法的测定值基本一致。
本工作采用溶液法制样,以锶内标法建立了XRFS测定天然铀产品(U3O8)中铀含量的方法,并以标准曲线参数建立的校正公式消除了基体效应,所得结果的准确度和精密度均较好,可以满足天然铀产品(U3O8)贸易中铀的检测要求。
