原发性干燥综合征合并帕金森综合征五例临床分析
2021-10-08魏杰毛薇许二赫
魏杰 毛薇 许二赫
原发性干燥综合征(primary Sjögren syndrome,pSS)是以眼干、口干为主要特征的侵犯外分泌腺体的一种慢性自身免疫性疾病,除累及外分泌腺体外还可累及腺外组织,如累及神经系统者约占20%,其中累及中枢神经系统较为少见,且其临床表现也多种多样,主要包括中枢性脱髓鞘样改变、无菌性脑膜炎、头痛、共济失调、帕金森综合征、视神经脊髓炎、认知障碍和抑郁等[1-2]。其中合并帕金森综合征者更为少见,国外于1993年由Visser等[3]报道了第一例pSS合并帕金森综合征的病例,国内最早于2011由周志华等[4]报道了一例,随后陆续有相关病例报道,但患者临床表现、影像学特点及对左旋多巴制剂、糖皮质激素等治疗的反应均不同。本研究通过分析5例pSS合并帕金森综合征患者的临床资料,并结合文献进行总结,旨在探讨pSS合并帕金森综合征的临床特点及可能发病机制。
1 对象和方法
1.1 对象收集首都医科大学宣武医院2016年6月至2019年7月诊治的pSS合并帕金森综合征患者5例,所有患者符合pSS诊断标准及帕金森病诊断标准中的帕金森综合征的诊断[5-6]。
1.2 方法回顾性分析5例患者的临床表现、实验室检查、影像学特点、药物治疗及疗效资料。5例均行颅脑MRI检查,3例行腮腺动态核素显像,1例行唇腺活检,2例行PET-CT检查,1例行AV-133-PET/CT检查,3例行美多芭药物负荷试验。所有患者行血常规、红细胞沉降率、风湿三项+免疫五项、甲状腺功能、肿瘤标志物、抗核抗体谱、抗中性粒细胞抗体、抗心磷脂抗体、抗β2-糖蛋白1抗体及自主神经功能等检测。
2 结果
2.1 临床特点5例患者中女性4例,男性1例;发病年龄平均65岁;2例单侧起病,表现为偏侧肢体震颤及强直,3例双侧起病,表现为对称的肢体强直及步态不稳等症状;1例帕金森综合征症状出现于pSS症状之前,3例出现于之后,1例几乎同时出现临床症状。结果见表1。
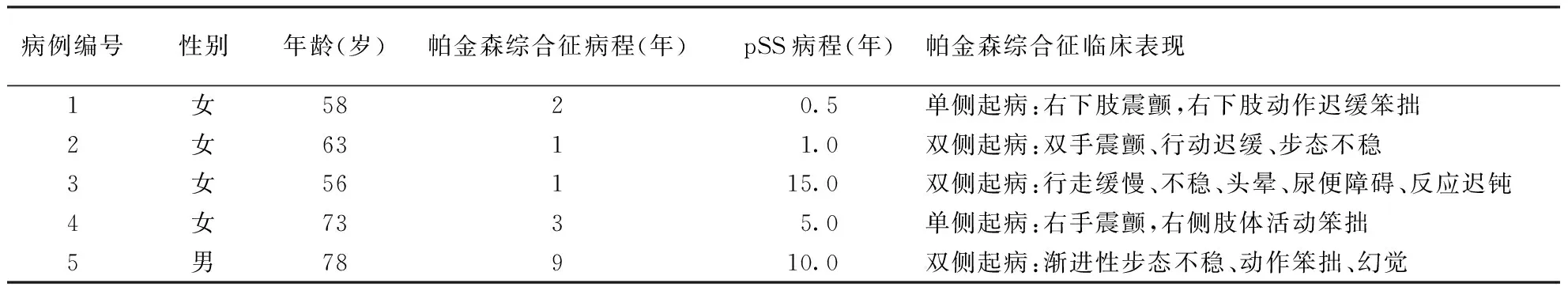
表1 5例pSS合并帕金森综合征患者的临床特征
2.2 实验室检查5例患者血常规、红细胞沉降率、风湿三项+免疫五项、甲状腺功能、肿瘤标志物、抗中性粒细胞抗体、抗心磷脂抗体、抗β2-糖蛋白1抗体检测结果未见异常。其中3例抗SSA抗体阳性,4例抗Ro-52抗体阳性,1例抗SSB抗体、抗Ro-60阳性,1例抗CENP B抗体、抗线粒体抗体M2型抗体阳性(表2)。3例行腰穿检查,脑脊液免疫球蛋白A、M、G均升高,2例24 h IgG鞘内合成率明显升高。
2.3 美多芭药物负荷试验3例给予美多芭250 mg行药物负荷试验,对美多芭反应较敏感,另两例因各种原因未行此试验。具体结果见表2。
2.4 影像学检查3例头颅MRI检查显示基本正常或有小的腔隙性梗死病灶,但未见明显脑白质病变(图1、2),2例存在脑白质病变(图3),其中1例可见小脑、脑干萎缩(图4),病例1行FDG-PET/CT检查可见双侧尾状核头部代谢轻度减低(图5),AV-133-PET/CT检查可见左侧壳核放射性分布减低(图6)。病例2行PET-CT检查显示右侧额叶和颞叶代谢轻中度减低,双侧小脑半球代谢中度减低,以右侧为著。具体结果见表2。
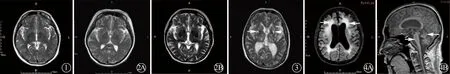
图1 病例1头颅MRI T2像可见颅内有少许缺血灶图2 病例3(A)和病例4(B)头颅MRI T2像未见明显白质脱髓鞘图3 病例2头颅MRI T2像可见脑内多发腔隙性脑梗死,脑白质变性(箭头所示)图4 病例5头颅MRI Flair像可见明显白质髓鞘表现及脑萎缩(A,箭头所示),矢状位可见明显小脑及脑干萎缩(B,箭头所示)
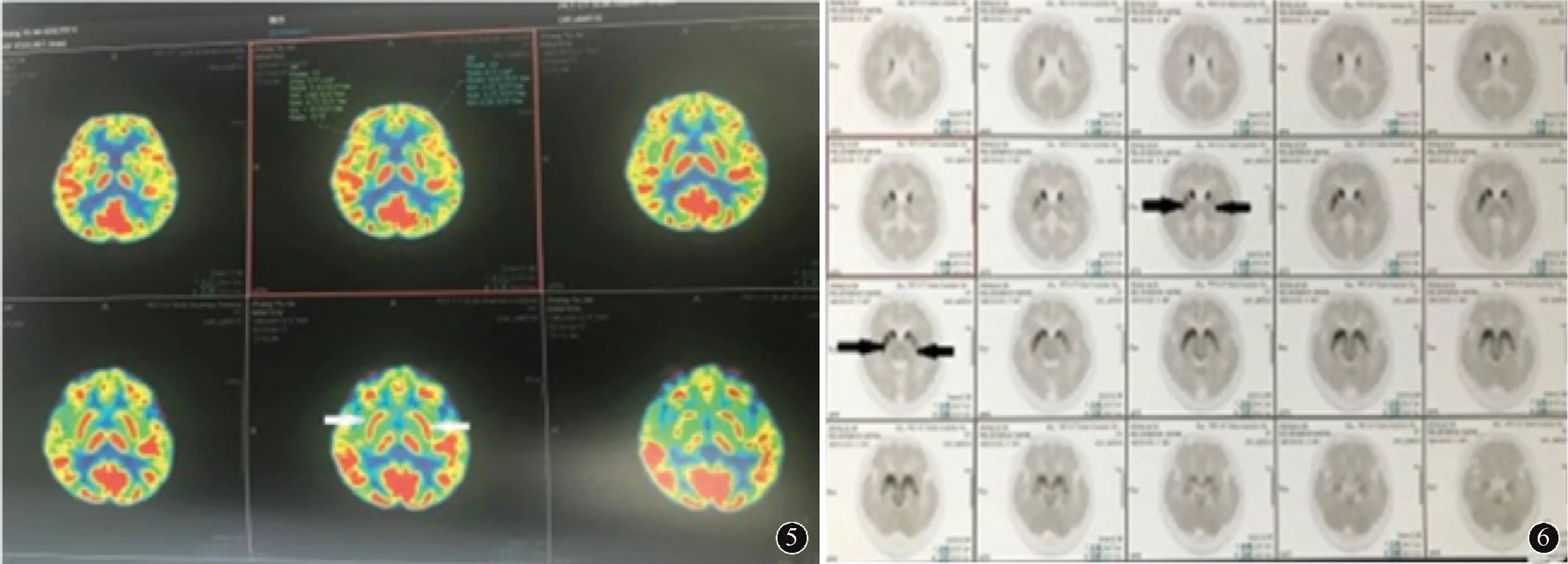
图5 病例1 FDG-PET检查示双侧尾状核头部代谢对称,稍减低(箭头所示)图6 病例1 AV-133-PET检查示左侧壳核放射性分布减低(箭头所示)
2.5 腺体相关检查4例行腮腺动态核素显像检查,结果显示均存在腮腺摄取功能或分泌功能受损情况(图7)。1例行唇腺活检,可见涎腺小叶萎缩,间质内散在淋巴细胞和浆细胞浸润,可见含50个以上淋巴细胞浸润的聚集灶1个(图8)。具体结果见表2。
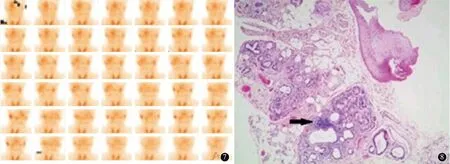
图7 病例1腮腺动态核素显像示右侧腮腺摄取功能较对侧减低图8 病例1唇腺活检示涎腺小叶萎缩,间质散在淋巴细胞及浆细胞浸润,见含50个以上淋巴细胞的聚集灶1个(箭头所示,HE ×80)
2.6 自主神经功能检测1例存在低位性低血压,2例膀胱残余尿量异常,1例肛门括约肌肌电图检查结果异常。具体结果见表2。
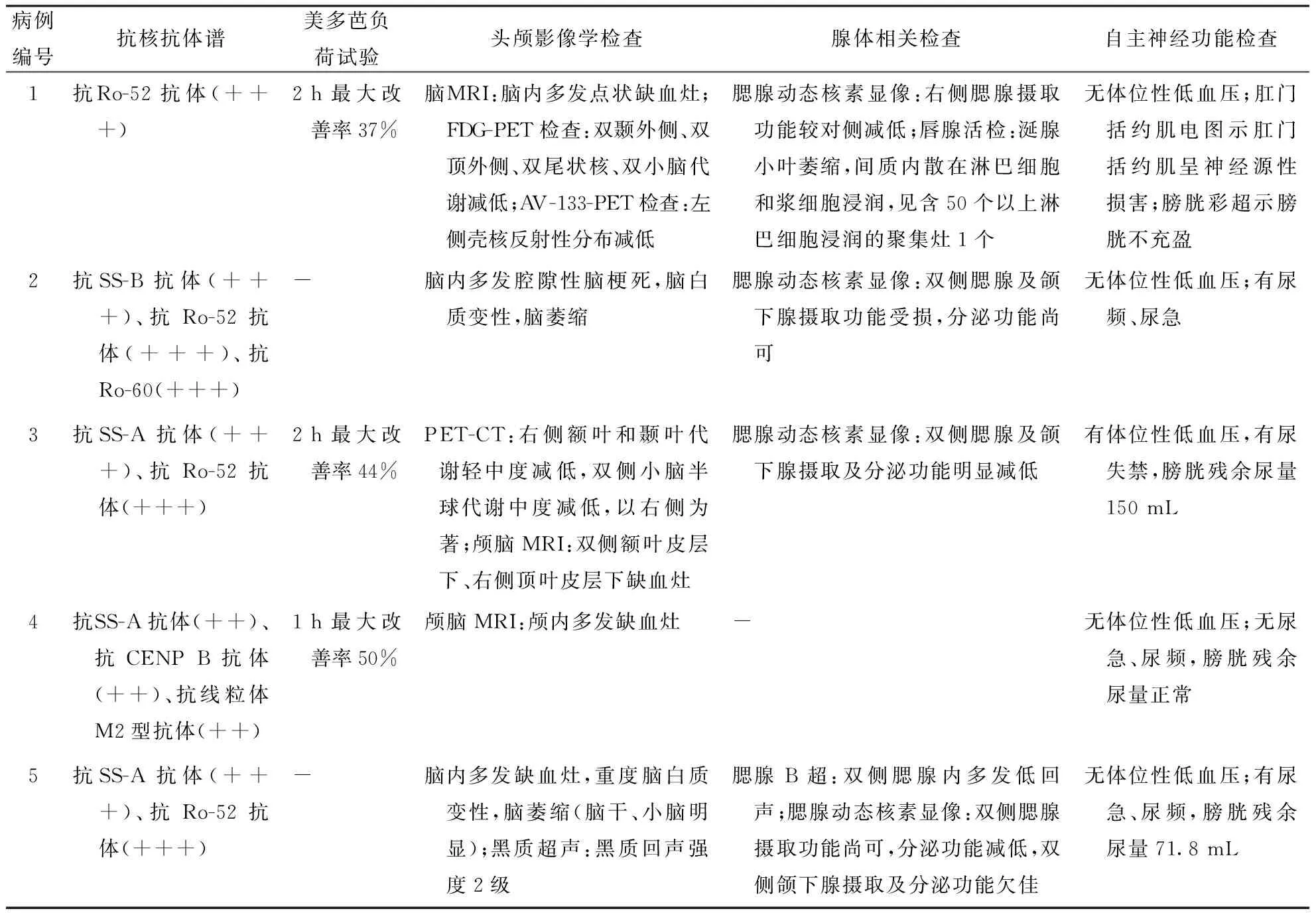
表2 5例pSS合并帕金森综合征患者辅助检查结果
2.7 治疗2例单纯以震颤起病的患者,最初被诊断为帕金森病,仅给予抗帕金森病药物,效果不理想,后诊断pSS后加用糖皮质激素或免疫调节剂后症状改善。3例起始给予糖皮质激素治疗,后期均给予联合治疗,1例药物效果较好,1例效果尚可,因病程较长,需进一步长期随访观察,1例效果差。具体结果见表3。

表3 5例pSS合并帕金森综合征患者治疗及疗效
3 讨论
该组5例患者中女性4例,男性1例,平均发病年龄65岁,与既往报道女性多发且平均发病年龄为61岁一致;2例为单侧受累,表现为震颤及强直,3例为双侧受累,表现为对称的强直及步态不稳等症状,这与既往报道的多为强直而不伴震颤的结果不同;1例帕金森综合征症状出现于pSS症状之前,3例出现于之后,1例同时出现;3例头颅MRI检查仅见小的腔隙性梗死病灶,未见明显白质脱髓鞘病变,2例有脑白质病变,其中1例可见小脑、脑干萎缩,这与既往报道不同,既往报道大部分患者头颅MRI检查显示脑白质、纹状体、苍白球部位T2及FLAIR高信号,小部分患者头颅MRI正常[7]。1例行FDG-PET/CT及AV-133-PET/CT检查显示存在代谢改变与帕金森病相同。检索既往病例报道未发现有Ⅱ型囊泡单胺转运体(vesicular monoaminetransporter-2,VMAT2)的相关影像学检查。3例行美多芭药物负荷试验,对于美多芭反应均较敏感,而既往报道显示大部分患者对多巴胺类药物抵抗。
pSS作为一种自身免疫性疾病,当累及到黑质-纹状体系统可导致震颤、强直、姿势平衡异常等帕金森综合征表现,上述症状大多出现于干燥综合征后数年。该组5例患者的帕金森综合征病程相对较长,部分病例进展较慢,而较多累及基底节区、小脑、脑干等部位,反映了其发病机制可能与经典的帕金森病、多系统萎缩等神经系统变性疾病不尽相同或包含多种机制。最初有部分学者认为由抗SSA或抗SSB抗体直接损伤基底节区神经细胞而导致出现锥体外系症状[8],随后Hassin-Baer等[9]发现3例pSS合并帕金森综合征患者的血清抗β2-GPI抗体均为强阳性,推测该抗体与抗原结合,在补体参与下形成免疫复合物,沉积在血管壁引起血管炎而出现帕金森综合征的临床表现。该组5例患者抗β2-GPI抗体均为阴性,不支持该推断。另一种假设为炎症介质如肿瘤坏死因子α(TNF-α)、白细胞介素-1β(IL-1β)、IL-6以及一氧化氮等激活大脑中的免疫细胞(小胶质细胞),可能促成黑质多巴胺能神经元的不可逆凋亡[10]。本组5例患者未发现相同的免疫相关抗体,由于未对5例患者行TNF-α、IL等炎症因子检测,是否支持以上的机制推测尚不清楚,但其中3例行腰穿检查发现均存在免疫球蛋白A、M、G升高,2例24 h IgG鞘内合成率明显升高,表明患者颅内存在免疫损伤,但具体通路仍需进一步研究。
本组患者中有1例帕金森综合征表现出现于干燥综合征之前,表现为强直少动、静止性震颤,并对多巴胺能药物敏感,无绝对排除标准及警示征,行AV-133-PET显示对应侧壳核后部代谢减低,根据2015年MDS帕金森病临床诊断标准,患者符合临床确诊的帕金森病,作者推测帕金森病的发病可能并非由pSS累及到黑质、纹状体系统后导致,而是两者可互相增加另一种疾病的发病风险。近年有研究[10-12]结果支持作者上述观点。黄燕等[13]于2007年报道了1例多系统萎缩合并干燥综合征的患者。本组中病例3反复住院,有小脑功能障碍,同时有体位性低血压、尿失禁,残余尿量150 mL,根据MSA诊断标准符合很可能的MSA;病例5有小脑功能障碍表现,同时有尿急、尿频,膀胱残余尿量71.8 mL,颅脑MRI可见小脑、脑干明显萎缩,符合诊断为可能的MSA,但该患者病程长达9年,且临床自主神经功能受损表现不突出,无体位性低血压表现,服用免疫抑制剂等治疗效果可,预后相对良好,这与典型的MSA预后不良不同,故推测也有pSS合并多系统萎缩的可能,但两例均未行VMAT2相关影像学检查。Heidary等[14]曾报道过pSS并发小脑、脑干萎缩的病例,这与本组上述两例相似。此外,亦有pSS导致自主神经功能障碍的病例报道,行心脏131I标记间碘苄胍(131I-MIBG)后明确有心脏自主神经受损,同时合并有体位性低血压及膀胱功能受损[15],但并未诊断为多系统萎缩,这为pSS可能合并多系统萎缩提供了线索。
综上所述,本研究结果显示,干燥综合征合并帕金森综合征患者均存在腮腺摄取或分泌功能不同程度损伤,帕金森综合征的临床表现多样,可同时伴有小脑性共济失调、自主神经功能障碍等表现。其可能的机制为通过多种炎症介质如TNF-α、IL等介导,损伤黑质处神经元,导致帕金森综合征的出现,pSS合并帕金森综合征可能为两个独立疾病,具体发病机制及通路仍需进一步探究。
