锂硫电池中的催化应用
2021-09-28高希雅邓子华李存璞魏子栋
高希雅,邓子华,李存璞,魏子栋
(重庆大学化学化工学院,重庆 400044)
1 锂硫电池基本原理与存在问题
可持续能源技术的蓬勃发展标志着现代社会的能源革命,探索可持续、清洁、安全的新能源供应已成为了国际共识。新能源电动汽车、便携式电子设备和移动电源等领域的兴起使得人们对储能设备提出了更高的要求。在热能、风能、机械能等多种储能系统中,具有高能量密度储能的可充电电池成为了最常用的电化学储能设备。其中锂硫电池以其高理论容量(1672mA·h/g)、丰富的自然资源和环境友好性等优点受到越来越多的关注,是新兴电池技术中极具吸引力和发展前景的候选技术,是满足全球日益增长的能源消费需求的理想下一代储能设备。
锂硫电池的充放电过程有溶解和沉积两个步骤,包含固相-液相-固相的相变转化。在放电过程中,正极的单质S8被多步还原成Li2S,放电曲线通常会出现两个平台,分别位于2.4V和2.1V左右,如图1所示。第Ⅰ阶段的放电平台为2.4V,对应单质S8开环并与迁移到正极的锂离子成键生成Li2S8的固-液两相还原过程和Li2S8向可溶性长链Li2S6转变的液-液单相还原过程;第Ⅱ阶段的电压从2.4V骤降为2.1V,可溶性Li2S6再还原生成可溶性Li2S4,且第Ⅰ、Ⅱ阶段共提供约25%的理论硫容量。第Ⅲ阶段的放电平台为2.1V,包含可溶性Li2S4向不溶的短链Li2S2转变的液-固两相还原过程和Li2S2向Li2S 转变的固-固单相还原过程,贡献约75%的理论硫容量[1]。在充电过程中,Li2S 首先转化为不溶的短链Li2S2,然后进一步生成可溶性长链Li2Sn(4≤n≤8,n为整数)(LiPSs),最后转化为S8。电极反应和电池总反应如式(1)~式(3)。

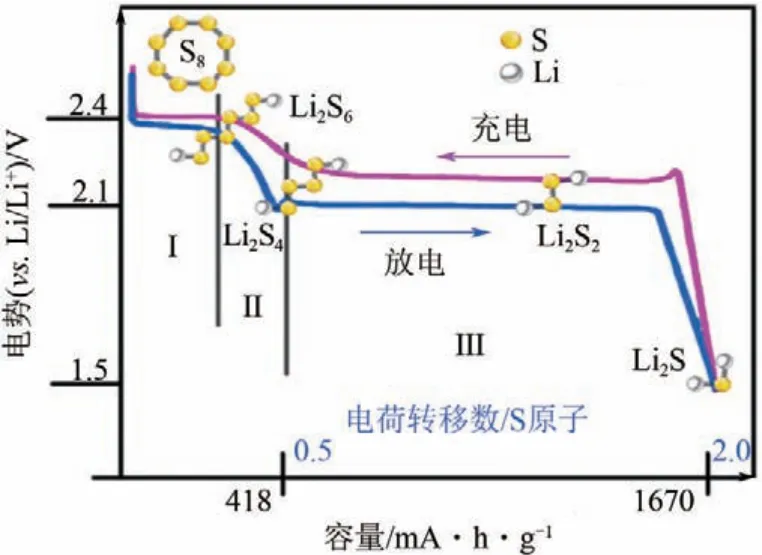
图1 锂硫电池的充放电曲线[1]
锂硫电池作为具有应用潜力的高能量密度储能系统,当前仍存在众多挑战,严重影响着电池的倍率性能和循环寿命,使其不能达到预期的理论水平,极大地阻碍了实际商业应用化进程[2-3]。具体问题如下:①单质S8和固体产物Li2S2/Li2S的导电性较差,限制了电子在硫阴极中的传递和活性硫的利用;②穿梭效应造成活性物质硫的不可逆损失,使得电池自放电现象严重,导致电池库仑效率降低,循环寿命缩短[4];③充放电过程中硫的体积膨胀,导致宿主材料的结构易粉粹和崩塌,影响电池的循环性能。
以上问题中,由不同浓度梯度的LiPSs 造成的穿梭效应最能影响锂硫电池的性能。在实际放电反应进程中,可溶性LiPSs 和不溶性Li2S2/Li2S 之间存在相的转化,且固体Li2S2的成核势垒较大,其成长速度大于成核速度,导致固体产物生成滞后,进一步导致可溶性LiPSs 在正极侧的堆积过剩。反观由于Li2S 具有较高的活化能和较差的导电性,使得Li2S在充电过程中难以完全再氧化,不均匀滞留在正极表面,降低了反应的可逆性和可循环性。因此,研究者们开始关注如何减缓穿梭效应的同时提高反应动力学,从而加快整个反应进程,提出了锂硫电池中的催化作用,认为推动可溶性LiPSs 和固态Li2S2/Li2S的快速互相转化,克服液固转化中的能垒是实现锂硫电池高能量密度的一条有希望的途径[5]。
2 锂硫电池催化材料
与可溶性LiPSs 的物理约束相比,化学吸附和催化转化表现出更好的抑制穿梭效应的能力[6-7]。本节总结了各种应用于化学吸附和催化转化可溶性LiPSs 的材料,包括过渡金属氧化物、氮化物、硫化物、碳化物、磷化物、金属有机框架、共价有机框架、导电性载体等。
2.1 过渡金属氧化物
过渡金属化合物(MaXb,M 为金属,X 为阴离子)是一种用途广泛的材料,因为它们的物理/化学性质取决于M和X。大多数过渡金属化合物具有适合与硫类化学反应的极性表面,因此它们占锂硫电池催化材料中的大部分[8]。过渡金属氧化物具有较大的电化学活性表面,并含有亲水性基团,是很好的硫宿主材料[8]。O 原子具有空d 轨道,是良好的电子接受体,容易和锂或其他金属化学键合,提高与可溶性LiPSs 的结合能,相互作用强烈,降低了Li2S 成核的动力学能垒,有利于Li2S 的沉积和分解。
王晓敏团队[9]提出以Fe3O4纳米粒子/分级多孔碳(Fe3O4/HPC)为阴极和FeP/HPC 改性隔膜的双功能策略,以提高LiPSs 的锚定和催化性能,保证均匀的Li2S沉积,降低死硫。结果表明,组装电池在1C的电流密度下循环1000次后,以0.083%极低的衰减率使得容量保持为562.4mA·h/g。系统的理论计算如图2 所示,由于Fe—S 键和Li—O 键的存在,p带中心发生了偏移,使得Fe3O4与Li2S4和Li2S6的结合能更强,加速了Li2S4和Li2S6的氧化还原动力学反应,提高了固体Li2S沉淀的均匀成核生长。
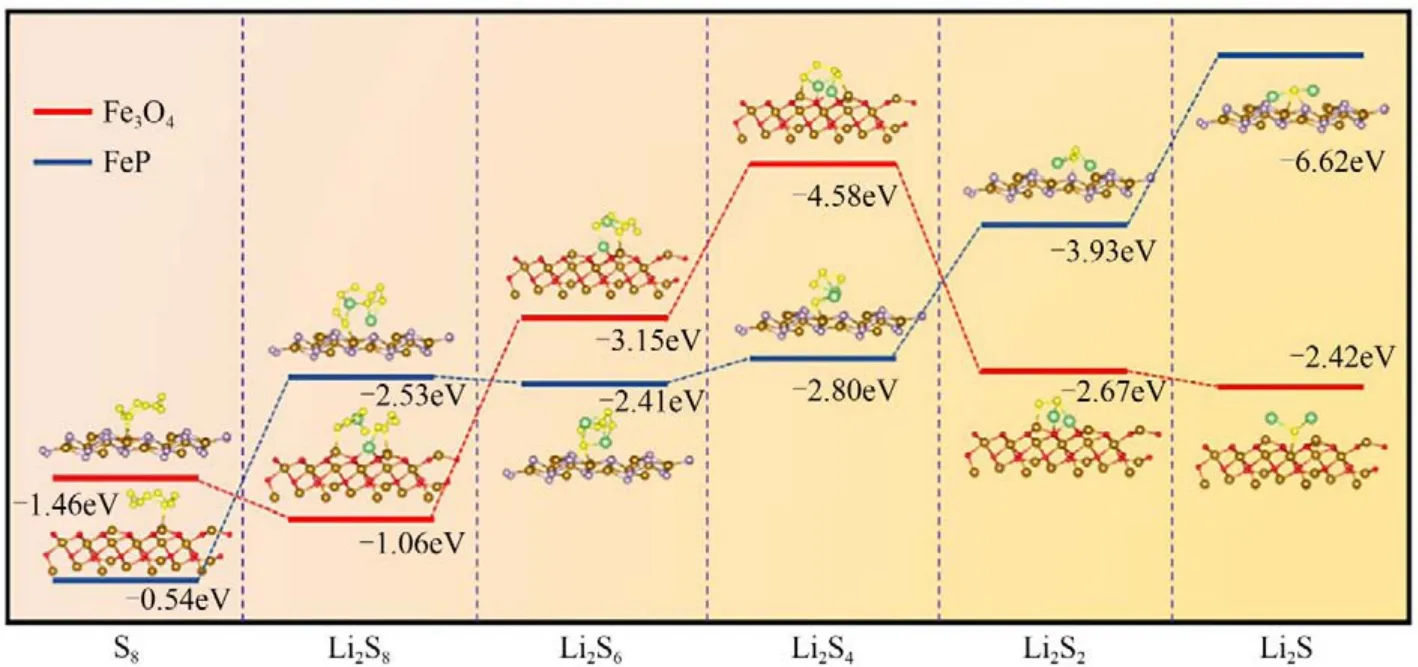
图2 不同反应产物的DFT计算模型和吸附能[9]
Nazar 等[10]用MnO2壳层包裹硫颗粒,利用KMnO4与硫之间的氧化还原反应。形成的纳米级MnO2壳层与可溶性LiPSs 在物理和化学上相互作用,并有效捕获它们,这使得1700 多次循环后还能保持0.039%的低容量衰减。Lu 等[11]研究了将单质硫固定在G/MnO2复合材料表面以提高硫电极的循环稳定性的方法。由于多孔石墨烯具有良好的导电性和MnO2的催化性能,电极为G/MnO2/S 的电池在0.2C和1C下的初始放电容量分别为1132mA·h/g和908mA·h/g。1C下完成500个循环后的保留率为43.78%以上。
Liu等[12]和Nazar等[13]分别证明MnO2和V2O5可以将LiPSs 氧化到高氧化态以达到限制中间产物的作用。高氧化还原电位的金属氧化物可以与LiPSs 相互作用形成硫代硫酸盐/聚硫酸盐表面物种,甚至通过表面氧化还原反应形成硫酸盐物种,从而抑制LiPSs 的迁移,提供锂硫电池的循环稳定性[14]。例如,LiPSs与MnO2之间的氧化还原反应可以发生在MnO2的表面[13]。随着Mn4+还原为Mn2+,MnO2表面的多硫离子被氧化,生成硫代硫酸盐基团。表面的硫代硫酸盐基团作为锚定位点,使LiPSs 形成聚硫酸盐并引发Li2S2/Li2S的转化。
CeO2[15]、TiO2[16]、WO3/ZrO2[17]等多种氧化物催化LiPSs 的转化均有报道。此外,双金属氧化物如Ni-Fe等[18]具有多催化位点而受到关注。
2.2 过渡金属氮化物
金属氮化物对LiPSs 的转化具有很强的催化作用,其高电导率是主要优势,它可以使活性硫快速传递,从而提高活性硫的电化学利用率。
李红团队[19]通过构建有氮化钴(Co4N)催化剂的双壳中空纳米笼提高了LiPSs氧化还原动力学。N掺杂的中空内碳壳不仅可以作为LiPSs 的物理化学吸收体,而且提高了电极的电导率,显著抑制了穿梭效应。Co4N 纳米粒子嵌入在N 掺杂碳的外壳中,催化LiPSs 的转化,在循环过程中极化减少,动力学加快。锂离子插层能量学的理论研究证实了与金属Co 催化剂相比,Co4N 催化剂的催化活性得到了提高。结果表明,该电极在5C时循环400次后容量保持为658mA·h/g,具有良好的稳定性。
Zheng等[20]合成了一种Fe2N@C纳米盒(Fe2N@CNBs)多功能硫主体,通过蚀刻与氮化结合的策略,用于高速率和长循环的锂硫电池。高导电性的碳壳物理上限制了活性材料,并提供了快速电子/离子传输的有效途径。同时,密度泛函理论计算和电化学分析证明了极性Fe2N核对LiPSs具有较强的化学键合和有效的催化活性。S/Fe2N@CNBs 电极即使在1C下循环600 次后,其容量仍保持在881mA·h/g,平均衰减率仅为0.036%,表现出高比容量、优越的倍率能力和长期循环稳定性。
氮化钛(TiN)表面与硫之间存在极强的键合,使得TiN 表面同时具有催化LiPSs 转化的活性和显著的锚定效应[21]。此外,TiN 有助于克服LiPS化学歧化反应迟缓的动力学问题。总的来说,TiN大大提高了锂硫电池的容量和速率能力,在5C下放电容量甚至达到700mA·h/g。除TiN 外,高导电性的InN[22]、VN[23]等也具有类似的电催化活性,被用于加快LiPSs转化反应。
2.3 过渡金属硫化物
近年过渡金属硫化物因其化学吸附能力和电催化作用而被发展成为理想的主体材料[24]。具有特殊离域电子结构的硫化物削弱了金属-硫键的离子性质,相比O2-来说,S2-是更软的碱,因而导电性得到了提升[25]。此外,硫化物的极性性质可以诱导LiPSs的快速转化,产生更多的活性位点[26-28],并促进Li2S的沉积行为,具有高催化效率的优点,进而提高活性硫的利用率,促进锂硫电池的反应动力学。
将纳米结构的VS4锚定在富含缺陷的碳纳米纤维(CNF)上[26],生成CNF-VS4并被包覆在商业隔膜上,作为电催化剂以增强LiPSs 生成Li2S 的反应动力学。极性纳米结构的VS4对LiPSs表现出较高的亲和力,导电的CNF网络作为“第二捕集剂”对活性物质进行捕获和再利用。功能涂层也为Li2S的分散和稳定提供了丰富的缺陷。当CNF-VS4功能隔膜基于高硫含量(质量分数80%)阴极时,在0.2C和2C 时的初始比容量也可以分别达到1135mA·h/g 和780mA·h/g。即使在更高的倍率5C 下,在经过1000 次循环后仍然提供约为300mA·h/g 的稳定容量。
Cao 等[27]利用原位合成的方法,通过CNTs 和ZIF-67 杂化结构制备均匀分散的Co3S4催化纳米颗粒,记为CNTs/Co3S4@NC。这种优化的结构使得硫和Co3S4纳米颗粒在ZIF-67 衍生物的N 掺杂碳纳米立方体内能够均匀分布和紧密接触,从而与LiPSs化学键合产生有效的相互作用,增强锂离子扩散,达到最大的催化转化效果,同时内置的三维碳纳米管网络确保了电极的高导电性。即使在高倍率5C下,也能维持约850mA·h/g 的比容量,循环1000次后保持85%的稳定性,对应平均每循环容量衰减率仅为0.0137%。
孙学良课题组[28]提出了一个独立结构的核桃状VS4纳米位点结合CNTs 作为阴极。在这种框架下,CNTs 阵列为高硫负载提供了高表面积和导电性,而VS4纳米位点有助于LiPSs 的捕获和催化转化。独立CNTs 阵列和VS4纳米位点具有高达6C 的高倍率能力,且VS4@CNTs/S 电极在2C 下以0.037%的低衰减率长期循环1200次。
王连洲团队[29]首次报道了通过原位聚多巴胺辅助硫化过程,在N、S 共掺杂多孔碳(TiS2@NSC)约束下,Ti3C2TxMXene 成功转化为三明治状超薄TiS2纳米片。作为硫的载体,即使在7.7mg/cm2高硫负载下,TiS2@NSC对LiPSs具有很高的捕获能力和显著的LiPSs还原和Li2S氧化的电催化活性。
崔屹课题组[30]对6 种金属硫化物(FeS、SnS2、Ni3S2、VS2、TiS2和CoS2)进行了研究。其中,VS2、TiS2和CoS2对Li2S 成核的动力学势垒较低,导致初始充电阶段电位丘急剧减小,这归因于锂离子在这些金属硫化物表面的高扩散率,容易电荷转移。因此,使用VS2、TiS2和CoS2的电极表现出稳定的循环寿命和高容量。这些研究表明,即使没有复杂的工程,金属硫化物也具有出色的催化活性。
2.4 过渡金属单原子催化材料
单原子催化是指稳定在合适载体上的孤立金属原子,可以提供高效和高活性的表面位置,从而可持续地催化转化。单原子催化不仅最大限度地利用了活性中心,而且提高了效率,减少了金属的使用,并提高了转化过程中的催化选择性。
美国韦恩州立大学Arava等[31]是第一批关注贵金属催化剂铂(Pt)对LiPSs氧化还原反应的催化活性的,他们将Pt催化剂纳米颗粒均匀分散在石墨烯层上,如图3(a)所示。含硫的Pt/石墨烯复合物电极在初始放电过程中的容量为1100mA·h/g,在100次循环后保持在789mA·h/g;优于初始放电比容量为740mA·h/g、在100 次循环后为580mA·h/g 的作为对照的石墨烯电极。为了考察Pt 的催化作用,从Tafel图中计算出交换电流密度(io),如图3(b)所示,Pt/石墨烯电极放电时io为3.18mA/cm2,石墨烯电极放电时io仅为1.18mA/cm2。在逆反应中,Pt/石墨烯电极的io同样高于石墨烯电极,说明Pt可在充放电过程中促进LiPSs的氧化还原反应。
金属纳米催化剂通过氧化还原过程促进了不溶态Li2S2/Li2S 向长链LiPSs 的转化,阻止了Li2S2/Li2S在电极上的沉积,促进了反应动力学。孙世刚课题组[32]发现,与纯石墨烯相比,Co@GC-PC的存在促进了放电过程中LiPSs的吸附,作用机理如图3(c)所示。密度泛函理论计算表明,Co 与Li2S 之间存在较强的相互作用,使得Li2S 在Co(111)上的分解势垒较低,有利于其在放电过程中的沉积和充电过程中的氧化。
另外,万立骏课题组[33]发现单分散Co 原子嵌入N掺杂石墨烯(Co-N/G)可以触发LiPSs的表面介导反应,使得S@Co-N/G电极在具有90%的高硫质量分数时,仍可以提供1210mA·h/g 的比容量以及硫载量高达6.0mg/cm2时,仍然具有每循环0.029%的低衰减率。图3(d)利用操作X射线吸收光谱和第一性原理计算相结合,证明了Co-N-C配位中心作为双功能电催化剂,分别促进放电和充电过程中Li2S的形成和分解过程。
张跃钢课题组[34]以原子分散的单个Fe 原子为催化剂,促进了相对惰性的Li2S阴极在长期循环过程中的脱锂过程,加速了可逆的电化学转化反应。12C下容量可达到588mA·h/g,5C下1000次循环的平均容量衰减率为0.06%,证明了单原子催化在提高锂硫电池性能方面的有效性。
2.5 碳化物与磷化物
碳化物中的过渡金属原子对LiPSs 的转化具有很高的催化活性,其表面具有较强的亲硫性,因此经常作为锚定材料来有效地吸附LiPSs。金属磷化物被认为是锂硫电池中最有前途的催化剂之一,由于亲脂相互作用形成P—Li键,促进硫代硫酸盐的形成,有利于短链Li2S2/Li2S的生成[35]。
Zhang 等[36]将Fe3C 纳米粒子嵌入N 掺杂多孔碳片中(Fe3C@NPCS),由于Fe3C 纳米粒子具有较好的电导率,可以通过快速电子传递加速LiPSs 的电化学反应,作用机理见图4(a)。另外,通过密度泛函理论计算证实,Fe3C纳米粒子能够通过强Fe—S化学键对LiPSs进行强吸附。
Yang 等[37]证明了磷化物FeP 能与LiPSs 形成较强的化学键,该纳米晶体对LiPSs 的转化反应和较低的Li2S成核能垒具有良好的催化作用。
此外还有B4C[38]、CoP[39]等材料均能表现出良好的性能和催化效果。
2.6 金属有机框架与共价有机框架
金属有机框架(MOFs)是由有机配体和金属离子或团簇通过配位键自组装形成的具有分子内孔隙的有机-无机杂化材料。具有较高的比表面积、不饱和的金属位点和良好的孔隙。共价有机框架(COFs)是一种新兴的多孔结晶框架,由轻元素(C、B、O 和N)组成,以共价键连接,具有规律的结构周期性、固有的孔隙率、结构的高度可调性和可设计性[40]。由强键构成的高度连接的晶体结构结合孔隙结构,使得构建单元为网状的MOFs 和COFs 的分子可以选择容纳高密度的固定阴离子,从而使锂离子成为唯一的可移动物种[41]。
董全峰课题组[42]合成了一种如图4(b)所示的新型的含Co 和N 掺杂石墨碳(Co-N-GC)的金属有机骨架衍生硫基体,具有大的比表面积,能够同时催化硫的氧化还原和LiPSs 的捕获,显著提高了锂硫电池的比容量、倍率性能和循环稳定性。
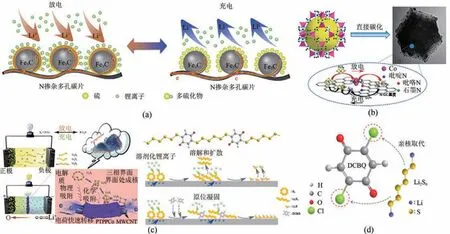
图4 Fe3C@NPCS复合阴极在充放电过程中的作用(a)[36]、双催化剂锚定Co-N-GC复合材料的制备及其与LiPSs的相互作用(b)[42]、PTPPCo/MWCNT修饰层对LiPSs协同抑制和催化的多功能机理(c)[47]以及DCBQ在Li-S氧化还原反应中的作用(d)[48]
Wang 等[43]提出了一种基于MOF(ZIF-67)衍生的原位掺氮碳纳米笼(CoP-HNC)作为高效硫载体。空心极性杂原子N掺杂碳结构具有丰富的孔隙和腔体,可有效缓解体积膨胀、缓冲电解质和捕获LiPSs。更重要的是,嵌套的极性CoP 纳米颗粒作为电催化剂,不仅锚定了LiPSs 中间体,而且能够显著促进LiPSs 转化的氧化还原动力学。CoPHNC 电极在5C时能提供约为800mA·h/g 的容量,1000 次循环后容量衰减率为0.02%,循环稳定性提高。
张永光课题组[44]以MIL-88B为例,提出将无定型金属有机骨架(aMOF)用于构建锂硫电池分离器。由于欠配位效应,aMIL-88B对硫类物质具有较高的吸附能力和催化活性,由aMIL-88B修饰的分离器实现高效和可逆的硫电化学,表现出极好的循环性,1C下循环500圈后仍有740mA·h/g的高容量保留。采用自模板配位-复制的方法开发了一种新型的三维有序宏观微孔金属有机骨架(3DOM ZIF-8)[45],作为提高锂硫电池性能的高级硫储层。独特的分层结构不仅有利于电解液的渗透和质子的传递,而且增加了活性界面大量暴露的表面积。此外,纳米ZIF-8 亚基与LiPSs 化学相互作用后对硫具有很强的固载和催化作用,从而能够明显抑制穿梭效应,并提高反应动力学。得益于这些协同特性,基于3DOM ZIF-8的硫电极表现出优异的电化学性能,即循环稳定性较长,500次循环后容量衰减率为0.028%,在提高硫负载和限制电解液的条件下,具有较高的面积容量和良好的循环性能。
张强课题组[46]采用以聚苯乙烯(PS)为模板制备了卟啉有机骨架空心球(POF-HSs)。POF-HSs得益于其极性化学结构和空心球形形貌,通过化学吸附和物理约束的双重作用,充分缓解了LiPSs 的穿梭现象,成为了硫阴极的理想主体材料,赋予锂硫电池高容量和循环寿命长的优良性能。
张嘉恒等[47]通过真空过滤的方法将聚四苯基卟啉钴(PTPPCo)吸附在多壁碳纳米管(MWCNT)上,制备了功能性隔膜(PTPPCo/MWCNT/PP),MWCNT具有优异的导电性,并可以促进沉积在阴极上惰性硫的再利用。PTPPCo不仅为捕获LiPSs提供了丰富有力的化学吸附位点,而且在LiPSs 转化反应中发挥了优异的催化作用,机理如图4(c)所示。此外,PTPPCo/MWCNT 复合材料易被电解质渗透,有利于锂离子的快速扩散。结合MWCNT和PTPPCo 的优点,具有PTPPCo/MWCNT/PP 的锂硫电池可以通过物理约束和化学吸附的双重作用有效地捕获LiPSs,提高氧化还原转化动力学,从而实现高硫利用率和转化率、优异的倍率性能,且循环性能稳定持久,抗自放电能力增强。
2.7 其他有机分子
吉林大学李峰课题组[48]采用原位凝固策略,通过2,5-二氯-1,4-苯醌(DCBQ)在电解质中引发亲核取代反应来有效地阻塞LiPSs。LiPSs 可以被DCBQ以固体有机硫的形式共价固定,使其有效地保留在阴极内,有助于提高容量保留率。此外,DCBQ中的苯并醌基能够加速锂离子的传递,促进硫的氧化还原反应动力学,其机理见图4(d)。
Goodenough 团队[49]利用双(4-硝基苯基)碳酸酯作为有机液体电解质的添加剂与可溶性LiPSs 反应生成不溶的Li2S2/Li2S 与4-硝基酚锂;4-硝基酚锂与锂金属阳极反应,生成一个有益于锂离子传导的阳极钝化层。
Wei 等[50]将吗啉分子成功接枝到商用聚丙烯分离器上。密度泛函理论(DFT)计算结果表明,吗啉侧链能均匀、可逆地吸附所有高阶聚硫化合物。这种双原子化学吸附调节了硫相关化合物之间的转化,加速了Li2S的成核过程。改进后的隔膜电池在0.5C下循环500次后,放电容量高达827.8mA·h/g,如图5所示。
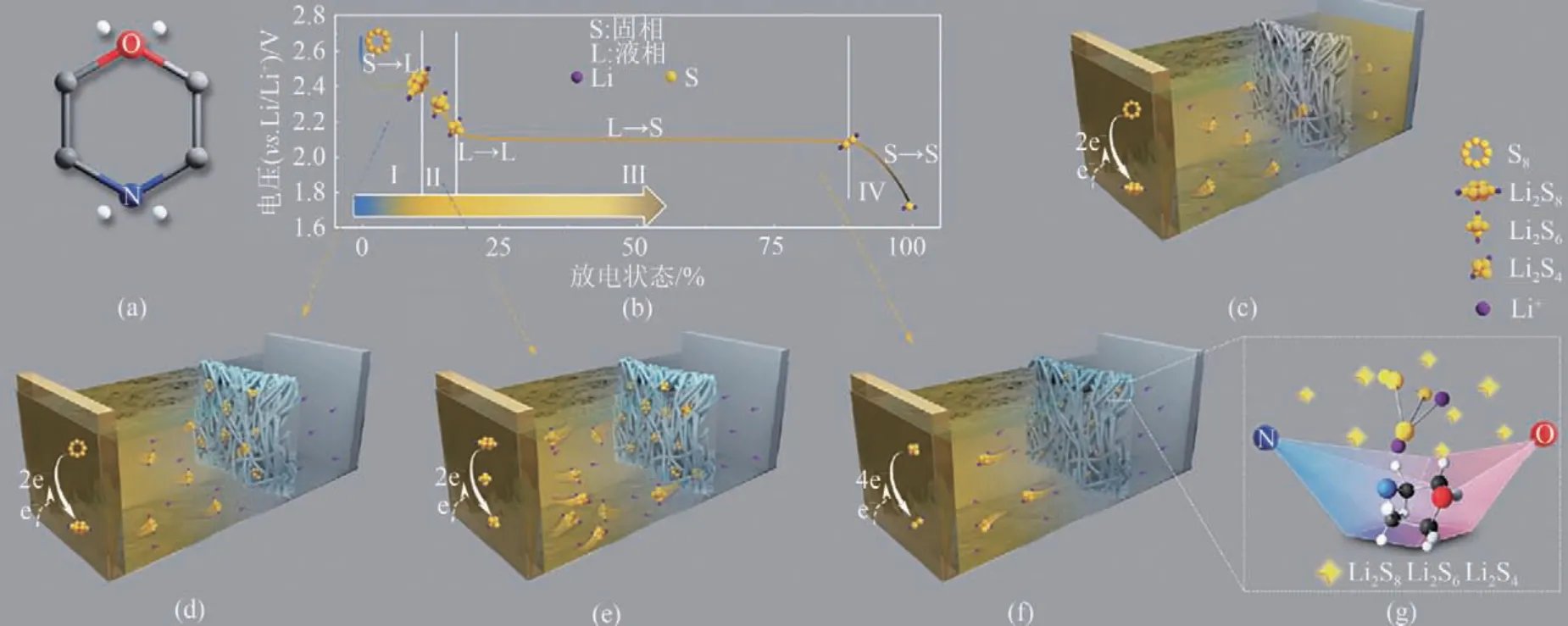
图5 吗啉的化学结构(a)、LiPSs在放电过程中对电势的演化(b)、PP分离器不能阻止溶解的LiPSs从阳极侧扩散的演示(c)、PP-C-St-MP分离器固定溶解的LiPSs并释放需要从第Ⅰ阶段到第III阶段在阴极侧转换时的LiPSs(d)~(f)以及吗啉的双原子化学吸附所实现的可逆吸附效应(g)[50]
另外,Wei 等[51]以“软硬酸碱理论”为基础,在商业隔膜上将三级胺覆盖层(TAL)与LiPSs选择性配位。用于捕获LiPSs 的锚定基团TAL 化学接枝到隔膜上,改变LiPSs 反应路径的同时又能保证有足够的孔,不会阻碍锂离子的传输,如图6 所示。因正电荷主要分散在团聚的LiPSs 分子的表面上,所以LiPSs是相对“软”的酸。当LiPSs进一步还原并释放到正极时,在隔膜上构建的“软”基TAL可以锚定LiPSs 软酸。而锂离子的尺寸小、电荷密度高,通常被认为是“硬”酸,它将被隔膜上的“软”基TAL 排斥。灵活的“多硫化物钳”构造方法为实现高能量密度锂硫电池铺就了一条新途径。
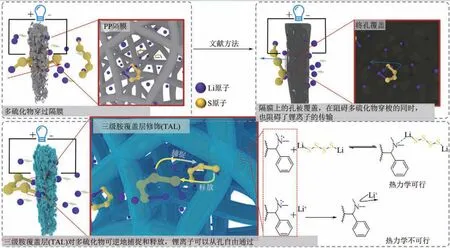
图6 锂硫电池隔膜的改性研究以及对LiPSs穿梭效应抑制机理[51]
2.8 导电性载体
张强课题组[52]通过热解生长在石墨烯上的二维卟啉有机框架(POF),制备了N 掺杂碳纳米片(PNG)用作中间层。由于POF的二维形貌,N元素完全暴露在石墨烯表面,而不受石墨烯骨架内电子传递的阻碍。原子表面被完全暴露的亲锂位点修饰,以提供对LiPSs的强化学吸附,并改善电解质的润湿性,而石墨烯衬底提供快速的电子传递,以促进硫物种的氧化还原动力学,如图7(a)所示。另外,他们还提出在还原氧化石墨烯纳米片(CoSe2/G)上原位生长钙矾石CoSe2纳米点,构建独特的电解质/CoSe2/G 三相界面[53]。该三相界面能够提供强的化学吸附、高的电导率和极好的电催化LiPSs 氧化还原反应。有效地增强了可溶LiPSs 的动力学行为,调节了大电流密度下固体Li2S沉淀的均匀形核和可控生长,如图7(b)所示。
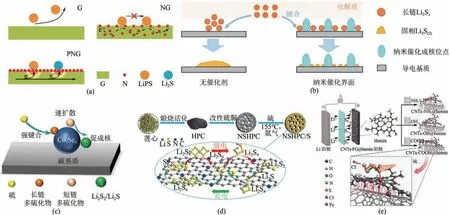
图7 PNG对LiPSs的作用(a)[52]、在常规导电表面和具有均匀形核位点的导电和催化纳米三相界面上的Li2S成核和生长(b)[53]、LiPSs在N-CN-750@Co3Se4-0.1mol·L-1基体上的吸附和转化(c)[59]、N,S共掺杂层次多孔碳/硫(NSHPC/S)复合材料的制备及与LiPSs相互作用(d)[61]、基于三种CNTs-FG@hemin阴极(FG=NH2,OH,COOH)的锂硫电池结构以及CNTs-COOH@hemin阴极上与LiPSs的作用机理(e)[63]
陈忠伟课题组[54]提供了一种利用天然丰富的有机分子蒽醌(AQ)抑制循环过程中氧化还原反应中LiPSs的溶解和扩散的策略。AQ很容易通过π-π相互作用与还原的氧化石墨烯(rGO)相连,随后与硫(S-AQ-G)混合。AQ 和LiPS 的酮基之间的强Lewis 碱-酸相互作用改善了氧化还原动力学,减少了极化。因此,S-AQ-G电极比硫与还原氧化石墨烯(S-G)复合电极具有更高的速率能力和循环稳定性。
3 催化性能提升策略
锂硫电池的实际应用还面临着诸多挑战[55]。单一策略很难达到锂硫电池长循环、低衰减的状态,为进一步催化转化多硫物种,采用双重策略如具有电荷效应(通过掺杂改变电子云密度)、吸附-催化共同作用(异质结)、双相催化作用(分解和沉积)等电催化材料已经被报道,它们不仅对LiPSs具有很强的亲和力,还可以极大地增强电化学反应动力学。
3.1 掺杂与功能化
长链可溶性LiPSs 和短链不溶的Li2S2/Li2S 末端硫均带有负电荷而具有相同的极性性质[56]。根据以往的研究发现,杂原子掺杂可以改变碳基体的表面电子结构,使非极化碳基体材料变为极化碳基体材料[57]。这些极性材料可以提供足够的电导率从而保证高硫利用率[35],其极性表面可以提供较强的亲和力。因而,掺杂被认为是一种有效的缓解穿梭效应的方法。其中,N掺杂碳材料是最常用的单掺杂碳材料。
张强课题组[58]分析了LiPSs与N、O、P、S、B、F 和Cl 等各种单掺杂原子之间的偶极-偶极静电相互作用,发现N或O掺杂的碳基体表现出增强的相互作用,可以阻止LiPSs 的来回移动。Han 等[59]提出了将高导电性的Co3Se4纳米粒子接枝到N掺杂3D碳基体表面制备的多功能硫基体,以抑制LiPSs 的穿梭,提高硫的利用率。通过调节碳基体和Co3Se4的分布,N-CN-750@Co3Se4-0.1mol/L 具有丰富的极性位点,对于LiPSs 吸收和硫的转化促进具有很好的表现,其机理如图7(c)所示。
除碳基单原子掺杂外,还经常采用双掺杂。对于双掺杂,两种杂原子在改善LiPSs 的电化学反应中起着至关重要的作用。大多数双掺杂碳材料都含有N 和B、P、S 或O。余桂华课题组[60]将植酸掺杂聚苯胺水凝胶设计合成了氮磷共掺杂碳(N,P-C)框架,作为无黏结剂的Li2S/N,P-C 阴极。N 和P共掺杂碳的三维多孔结构为锂离子的输运提供了连续的电子通道和分层的多孔通道。磷的掺杂还可以通过硫与碳骨架的强相互作用抑制穿梭效应,从而获得较高的库仑效率。与此同时,P掺杂在碳骨架中对改善反应动力学起着重要作用,它可以催化硫物种的氧化还原反应,降低电化学极化,提高Li2S的离子电导率。
王先友课题组[61]通过热解法和水热法成功合成了N、S 共掺杂分层多孔生物质碳(NSHPC)的蜂窝状结构,如图7(d)所示,这种双掺杂载体为溶解的LiPSs提供了化学吸附和活性位点。此外,N、S双掺杂还提高了多孔碳的亲水性和电导率,最终显著提高了锂硫电池的硫利用率和长期循环性能。
此外,Liu 等[62]研究了一系列金属/N 掺杂的半石墨有序介孔碳(Me-N-GOMCs,Me=Fe、Co、Ni和Cu)被设计为具有丰富孔隙率和高导电性的硫基质。结果表明,Fe 和N 的掺入可以显著地增强LiPSs的固碳性能。此外,将Fe-N-GOMC/S复合材料植入碳纸(CPs)的空隙空间内,构建了具有连续导电三维网络结构的独立集成硫阴极。快速的离子/电子传递和氧化还原动力学使锂硫电池在高负载下具有良好的硫利用率。结果表明,在0.5C下,CP/Fe-N-GOMC/S电极的初始容量高达1473mA·h/g,且在500次循环后容量衰减率可达0.075%。
杨植课题组[63]将血晶素(hemin)分子接枝到用官能团改性的碳纳米管(CNTs-COOH、CNTs-OH 和CNTs-NH2)上,合成了新型仿生分子催化剂并应用于锂硫电池正极材料中。其中,CNTs-COOH 与hemin 形成p-p 共轭和配位键,能提高硫的导电性和分散性,加之通过Fe—O 键配位的Fe(III)络合物对LiPSs 具有很高的吸附能力,可促进LiPSs 的快速转化,并能有效抑制穿梭效应,见图7(e)。
苏宝连课题组[64]通过亲核攻击与含氧官能团连接的几种碳原子来修饰带有乙二胺基团的碳纳米管网络。交联胺化碳纳米管框架为活性材料的快速电荷传递提供了途径,增强了活性材料的反应动力学,大大提高了活性材料的反应速率和硫的利用率。通过密度泛函理论计算证实,乙二胺基团的引入促进了表面极性的增强,从而有效地防止LiPSs溶解,提高循环稳定性。外部聚苯胺层从结构上抑制聚硫化物,以防止穿梭效应和活性物质损失。
3.2 异质结
金属碳化物、氮化物和磷化物是理想的介质材料,具有优越的导电性,有助于表面电荷传输和加速氧化还原反应。同时具有适度的表面极性,以平衡表面结合和扩散。但与LiPSs 的结合能相对较低,其吸附能力受到影响;而表面扩散缓慢的金属氧化物与LiPSs 具有较高的结合能。利用二者的结合构建高吸附性异质结构的材料,可以有效地提高LiPSs 的整体吸附性。特别是由于MXene 具有奇特的二维特性、极好的金属导电性和丰富的表面可调功能,是一种理想的异质介质[25]。
Nazar等[65]证明当表面反应性的—OH基团被硫取代时,MXene 能够通过S—Ti—C键与硫键结合;Ti2C表面的电子转移加速了表面氧化还原反应。据此提出了两步作用机制:端位—OH的MXene 相首先发生—OH 与LiPSs 的氧化还原反应,形成表面硫代硫酸盐基团,通过Wackenroder 反应进一步暴露Ti原子,然后亚稳Ti原子与额外的LiPSs通过路易斯酸碱相互作用反应生成S—Ti—C/N 化学键,如图8(a)所示。
杨全红课题组[66]通过原位制备TiO2-MXene(Ti3C2Tx)异质结构,提出了一种对LiPSs具有强吸附能力和优异转化活性,具有高比表面积和电子导电性等优点的多功能催化剂,如图8(b)所示。在MXene 表面均匀分布的TiO2作为俘获中心固定LiPSs,其异质界面保证了锚定的LiPSs从TiO2快速扩散到MXene,并赋予了端氧的MXene 表面高催化LiPSs 转化的活性。另外,他们还证明了氧化层的掺杂能够提高氮化物的催化活性[67]。硫掺杂的氮化钛(TiN)氧化层,形成的Ti—S 键能够从导电的Ti—N 基体上传递电子,从而保证了高催化活性。Ti—S 与Ti—O 键在原子水平上的结合有助于同时实现LiPSs的强捕获和快速转换。
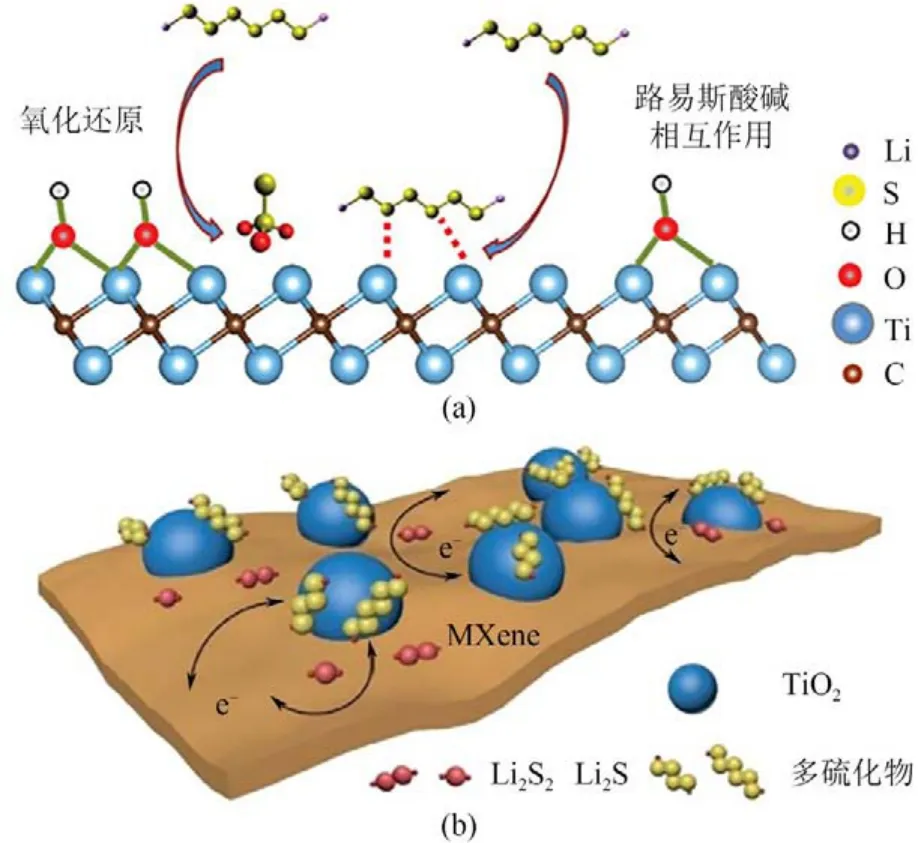
图8 带—OH的Mxene与LiPSs的相互作用(a)[65]以及TiO2-Ti3C2Tx结构上的LiPSs捕获和转化(b)[66]
Cheng 等[68]将 石 墨 烯@Mo2C (GCF-G@Mo2C)异质结构的三维分层石墨泡沫碳作为锂硫电池的独立电极。N掺杂泡沫碳被具有类似木耳的多孔结构的石墨烯片包裹,可以促进电子和离子的快速传递,同时通过强极性相互作用的物理约束和化学吸附有效地锚定LiPSs。此外,如图9 所示,Mo2C 纳米粒子可以作为化学锚定中心提供强的LiPSs 吸附,其催化作用加速了LiPSs 转化的氧化还原动力学,有助于提高速率性能。
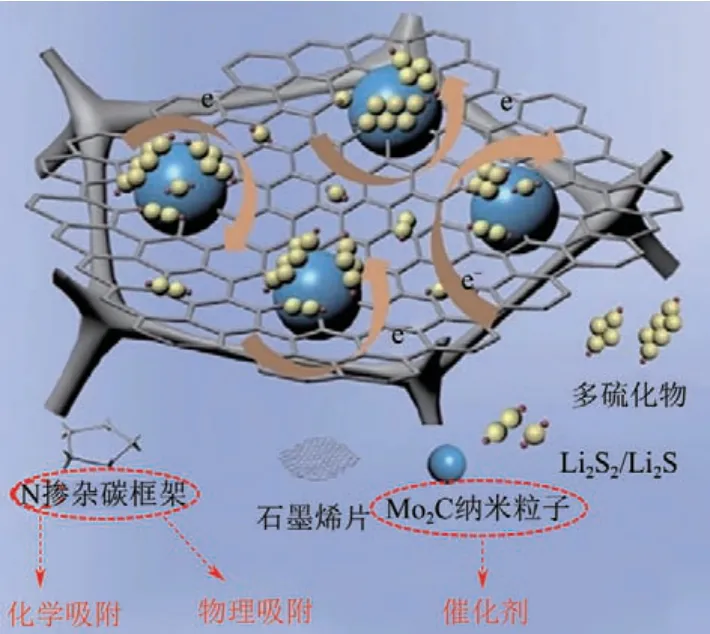
图9 GCF-G@Mo2C电极的制备过程及与LiPSs作用机理示意图[68]
吉林大学李峰课题组[69]将极性导电材料MoC@MoOx用于在氧化还原反应中LiPSs 的固定化和转化。MoOx的极性壳层能够对LiPSs进行化学吸附,MoC 的导电核有助于可溶性LiPSs 向Li2S 的转化。将MoC@MoOx材料掺入碳纤维泡沫电极中,电化学性能显著提高,具有较高的比容量和良好的循环稳定性。
4 催化剂评价方法
4.1 循环伏安法
循环伏安法(CV)作为最基本的电化学研究方法之一,测试电池的极化电流与电极电势之间的关系,可用于研究电极反应的性质、机理和反应动力学等。其原理是通过控制和改变电位以得到氧化还原电流方向。锂硫电池中存在复杂的电化学反应,因而根据CV 曲线上的具体特征分析氧化还原过程的细节,深入了解其中机理至关重要。
结合锂硫电池中的充放电曲线,采用CV 曲线描述电化学氧化还原过程。图10(a)为典型锂硫电池中观察到的CV曲线[70]。根据元素硫的反应机理,2.4~2.2V 处的首个阴极峰对应着S8到LiPSs 的液固转化过程,在2.1~1.9V 的第二个阴极峰对应着LiPSs还原成Li2S2和Li2S[71],这两个还原过程对应着放电过程中的两个电压平台。在2.2~2.5V处的阳级峰对应于Li2S2/Li2S 氧化为LiPSs/S 的过程[72],阳级峰在这个范围内极有可能重合。
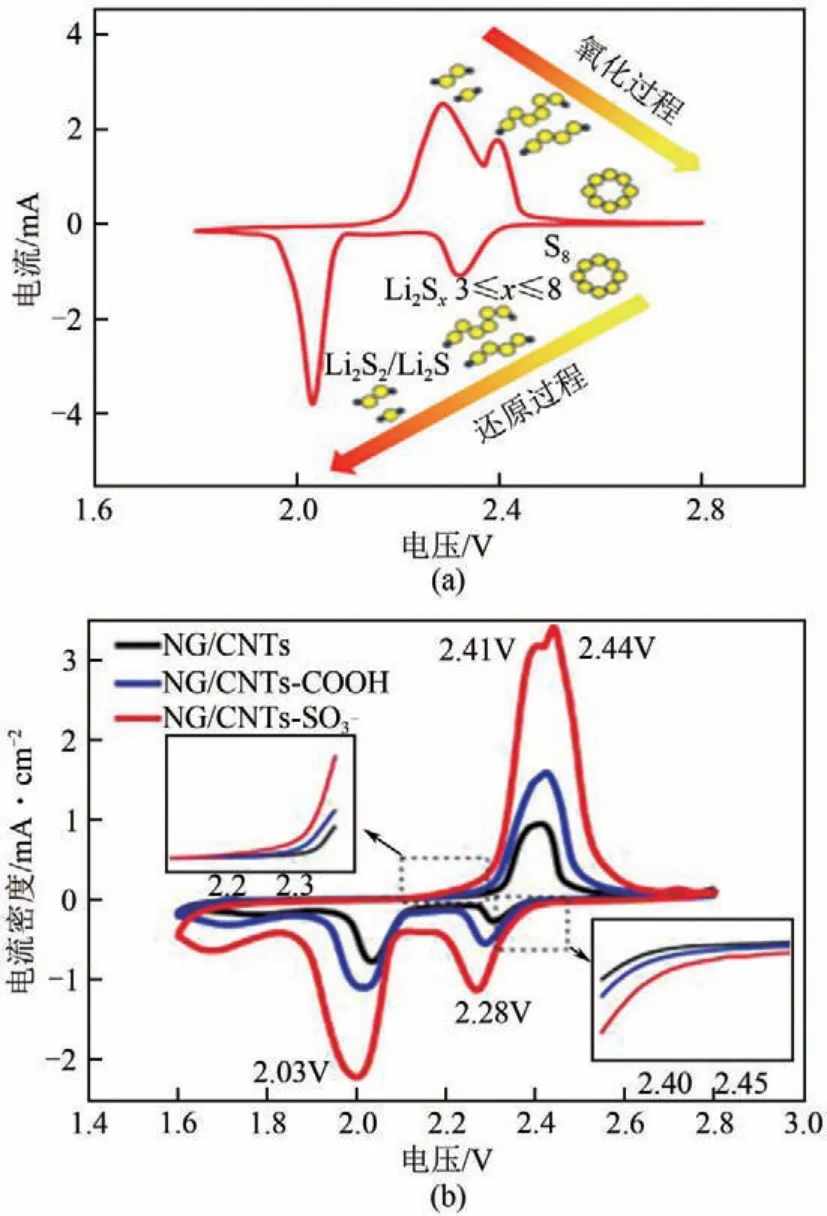
图10 锂硫电池中典型的CV曲线及NG/CNTs、NG/CNTs-COOH和NG/CNTs-S 电极的CV测试[70,73]
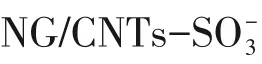
锂离子迁移系数(DLi+)可用于比较氧化还原反应动力学。CV测试中,电流(i)与不同的扫描速率的关系符合式(4)。

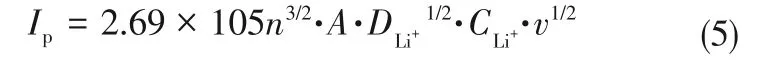

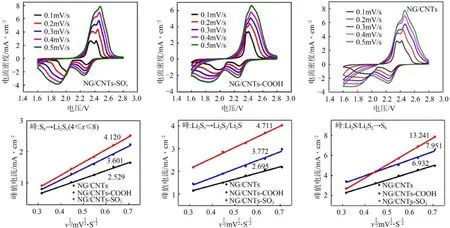
图11 NG/CNTs、NG/CNTs-COOH和NG/CNTs-S电极在不同速率下的CV测试及Ip随的变化曲线[73]
另外,可以采用对称电池的CV 测试评价反应动力学。对称电池由两个不负载硫的完全相同的电极组成,多以Li2S6为电解液添加剂。开路电压(OCV)设置为0,从OCV 扫描一个对称的电压区间,如-0.8~0.8V 或-1.2~1.2V。通过比较电流密度的大小,即可评价氧化还原动力学的快慢。
4.2 恒电流间歇滴定技术
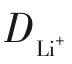
恒电流间歇滴定技术(GITT)[74]是一种暂态测量技术,由德国化学家Weppner提出。该方法假设扩散过程主要发生在固相材料的表层,具体来说,GITT 研究的是“物质的扩散过程与电荷转移”的关系,主要由两个部分组成。其中第一部分为小电流恒流脉冲放电,为了满足扩散过程仅发生在表层的假设,恒流脉冲放电的时间t要比较短,需要满足式(6)。第二部分为长时间地静置,以让Li+在活性物质内部充分扩散达到平衡状态。GITT 测试由一系列“脉冲+恒电流+弛豫”组成。

式中,L为材料的特征长度;D为材料的扩散系数[75]。
除了测定离子的扩散系数,GITT 常用于对电极反应中的微观动力学信息进行测定,在电化学方法与原理的基础上侧重分析电极和样品的化学反应与极化,以此为依据对不同充放电深度的极化及其可能发生的化学反应分段研究,逐步分析各阶段的差异,从而找到各阶段影响极化的关键因素。GITT最大的优势在于测量样品的真实度较高。
4.3 原位XRD技术
原位表征技术能够提供关于结构演化、氧化还原机理、固-电解质界面相(SEI)形成、副反应和锂离子输运的信息。原位XRD 通常用于监测在循环过程或温度变化过程中的材料的结构变化[76]。
Waluś等[77]通过原位XRD 技术检测锂硫电池的充电和放电过程,如图12(a)、(b)所示。发现最初存在于电极中的所有单质硫都被还原为可溶的LiPSs。在两个放电平台之间的区域,活性物质仍以可溶性形式存在于电解质中且未检测到峰。Li2S信号开始出现于2θ=11.61°、13.41°、19.01°和22.31°,分别对应于反射(111)、(200)、(220)和(311)面。Li2S 信号强度逐渐增大,直到在完全放电状态下达到最大值。这些宽峰意味着晶体太小或结晶不良,但都很好地集中在Li2S化合物预期的位置上。在充电过程中,Li2S 的强度逐渐降低,直到完全消失,证明了Li2S 可逆氧化为可溶的LiPSs。然而,由于Li2S 的绝缘性和不溶性,该化合物在电极中一直存在到高电荷状态(约74%)。在2.5V电荷平台的末端[图12(d)],非常明确的单质硫峰再次出现,它们的强度逐渐增加,直到电池充满电。
原位XRD 谱图中等高线图[78]明确地显示了硫依次还原为Li2S8、Li2S6,然后是Li2S4,最后是Li2S相,见图12(e)。此外,LiPSs 特别是长链LiPSs 的峰在整个循环过程中从未完全消失,导致了比容量的一小部分损失。令人惊讶的是,与常规的锂硫电池相比,未反应的长链LiPSs 并没有穿梭到锂阳极,也没有进一步反应,而是被应用的玻璃纤维分离器吸附。
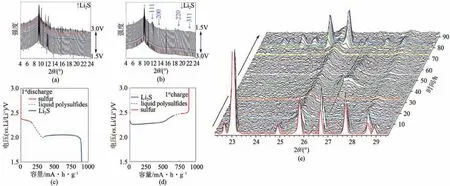
图12 锂硫电池在放电(a)和充电(b)过程中的XRD谱图及放电(c)和充电(d)的电化学图[77]以及锂硫电池的原位XRD测试(e)[78]
4.4 自由基检测

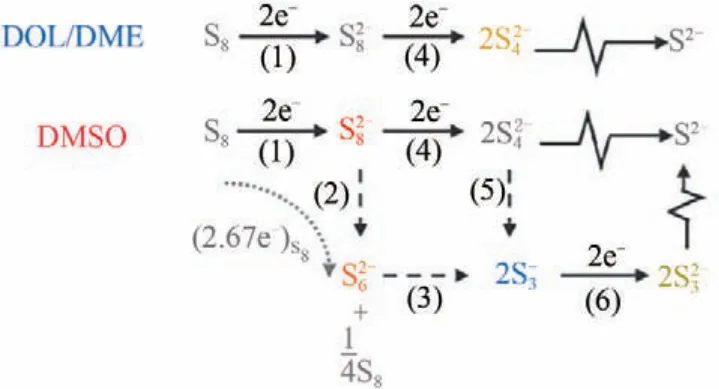
图13 提出的依赖溶剂的硫还原反应路径[81]
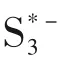
5 结语
对于锂硫电池来说,可溶性LiPSs 与不溶的Li2S2/Li2S 之间的转换缓慢是导致LiPSs 在电解质中穿梭的主要原因,严重降低了电池的能量效率和循环性能。因此,理想的硫载体不仅需要有很强的亲和力对LiPSs 进行锚定,还需要有推动LiPSs 向Li2S2/Li2S转化、有效加速Li2S成核和分解的催化能力。为加速氧化还原动力学,促进Li2S沉淀的均匀成核生长,多种催化材料已逐步应用于锂硫电池中。其催化机理可总结为以下几个方面:①导电空间限制多硫化物的迁移,提升多硫化物利用效率;②增大催化材料的极性与活性位点的暴露,吸附多硫化物,加速成核过程;③化学键合作用锚定催化材料与多硫化物,改变多硫化物沉积-还原路径;④提高电导率,加快电子传递,促进活性物质快速转化;⑤生成硫自由基,利用均相化学反应形成旁路反应机理。
通过催化转换这一策略抑制多硫化物的穿梭、促使多硫化物的快速转化,是锂硫电池未来的研究重点和电化学高密度储能领域中的关注热点之一,对锂硫电池这一高效的储能装置发展具有重要意义,也将帮助推进“碳达峰、碳中和”目标的实现。
