基于生物信息学分析免疫紊乱在类风湿关节炎发病机制中的作用▲
2021-09-22杨业静杜勇军李兴艳
杨业静 杜勇军 刘 雷 李兴艳
(广西南宁市第二人民医院关节骨科,南宁市 530031,电子邮箱:30764656@qq.com)
类风湿关节炎(rheumatoid arthritis,RA)为自身免疫性疾病,多累及小关节,以关节炎症、肿胀、疼痛为临床表现,女性多于男性,多高发于50~65岁人群。RA的全球患病率为0.5%~1.0%,我国的患病率约为0.42%[1]。近几年,RA患者因并发感染而住院的比例逐渐增加。一项对1993~2013年美国RA患者的大样本多中心流行病学调查显示,因感染住院的RA患者,从90/10万人增至206/10万人[2]。RA是威胁人民群众身体健康的重大疾病,也是心血管疾病的危险因素。但是RA的病因病机仍不十分明确,其发生与关节状态、饮食、代谢、家族遗传史等因素均有关。此外,越来越多的证据表明免疫紊乱参与了RA的发病,有学者在RA患者的滑膜组织中观察到白细胞介素(interleukin,IL)-1、IL-4等炎症免疫因子的浸润,以及自身抗体的沉积[3-4]。但是由于免疫系统疾病的复杂性,任何单一的假设均不能较好地解释免疫紊乱在RA发病中的作用机制。
生物信息学是一门新兴的学科,它将计算机与自然科学相联系,能用于解释复杂的生物机体,基因组学、代谢组学、蛋白质组学等均属于生物信息学范畴,该学科的发展为了解生理病理变化提供了新的思路。GEO数据库是用于储存芯片及测序数据的一个数据库,利用现有的公共生物数据库能整合多组不同实验室的结果,更好地挖掘潜在的信息。因此,本研究利用公共数据库,并联合多个RA滑膜组织的芯片,分析RA的差异表达基因以及免疫相关基因,进一步筛选T、B细胞的关键基因,并利用单细胞测序数据验证关键基因的表达,以探讨免疫紊乱在RA发病中的作用机制,并筛选小分子药物以干预RA。
1 资料与方法
1.1 数据来源 从GEO数据库(http://www.ncbi.nlm.nih.gov/geo)下载人类RA滑膜组织基因芯片数据,共有3个RA基因芯片数据纳入研究,其基本信息见表1,纳入标准:样品组织来源于滑膜,并同时包含RA的滑膜组织及正常对照组的滑膜组织。同时,下载RA小鼠外周血单细胞测序数据(GSE141105)。

表1 RA基因芯片信息
1.2 差异基因分析 首先对3个RA数据集的基因表达量进行log2校正以及基因名称注释。对数据进行批次校正联合分析后,利用R语言中的limma数据包分析基因芯片中差异表达的基因,以adj.P≤0.05、|log2FC|≥1为筛选差异表达基因的条件。利用R 语言中的火山图和热图可视化数据。
1.3 富集分析 利用R语言的clusterProfiler包对得到的差异表达基因进行京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析以及基因本体(Gene Ontology,GO)富集分析。以P<0.05表示富集的通路有统计学意义。
1.4 权重基因共表达网络分析 权重基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)通过邻接矩阵、拓扑重叠矩阵等方法,对相关性值进行幂次运算,从而将基因划分不同的模块。利用R语言的WGCNA包进行WGCNA分析,以分析RA的差异表达基因中存在相似表达模式的基因,并筛选关键模块。
1.5 免疫相关基因及免疫细胞浸润分析 (1)通过GO富集分析的“immune system process”筛选RA差异表达基因中与免疫相关的差异表达基因;利用STRING10(http://www.string-db.org)构建免疫相关差异表达基因编码蛋白的蛋白-蛋白相互作用 (protein-protein interaction,PPI)网络,并使用Cytoscape软件的插件Cytohubb筛选免疫相关的关键基因。(3)通过浓缩分数、补偿矩阵校正等算法,计算经校正后的3个RA数据集的基因表达量,得到免疫细胞含量矩阵,进一步得出免疫细胞浸润比例[5],以P<0.05表示差异有统计学意义。利用Pearson相关性分析评价关键基因表达量与免疫细胞含量矩阵的相关性,以P<0.05表示差异有统计学意义。
1.6 关键免疫相关基因在免疫细胞中表达情况的验证 利用RA小鼠外周血单细胞测序数据集GSE141105进一步验证1.5中所得到的关键基因在T、B淋巴细胞中的表达情况。采用秩和检验比较单细胞测序数据集GSE141105中正常组和RA组淋巴细胞之间关键免疫相关基因的表达差异,以P<0.05表示差异有统计学意义。
2 结 果
2.1 RA相关差异表达基因的筛选结果 对3个不同数据集进行批次校正后,共获得差异表达基因632个,其中表达上调的基因369个,表达下调的基因263个,在热图中(见图1)显示了10个最显著的差异表达基因,包括蛋白酶体20S亚单位β 9(proteasome 20S subunit beta 9,PSMB9)、泡状过表达癌症促生存蛋白1(vesicular,overexpressed in cancer,prosurvival protein 1,VOPP1)、γ氨基丁酸A型受体相关蛋白1(γ-aminobutyric acid type A receptor associated protein like 1,GABARAPL1)、蛋白质sel-1同源物3(protein sel-1 homolog 3,SEL1L3)、磷脂酶C-γ-2(phospholipase C γ 2,PLCG2)、C-X-C基序趋化因子配体(C-X-C motif chemokine ligand,CXCL)13、免疫球蛋白重链常数μ(immunoglobulin heavy constant μ,IGHM)、免疫球蛋白κ常数(immunoglobulin κ constant,IGKC)、去整合素和金属蛋白酶结构域样蛋白脱溶素1(a disintegrin and metalloproteinase domain-like decysin 1,ADAMDEC1)、黑色素调节蛋白(melanoregulin,MREG)。其他差异表达基因包括Toll样受体7(Toll-like receptor 7,TLR7)、肿瘤坏死因子配体超家族成员11(tumor necrosis factor super family 11,TNFSF11)、C-C基序趋化因子受体(C-C motif chemokine receptor,CCR)5、C-X-C基序趋化因子受体3(C-X-C motif chemokine receptor 3,CXCR3)、CXCL1(|log2FC|分别为1.55、1.42、1.77、1.00、2.418,adj.P值分别为3.877×10-9、2.554×10-8、2.87×10-9、18×10-5、9.20×10-9)。

图1 RA差异表达基因的火山图(A)与热图(B)
2.2 富集分析结果 对632个差异表达基因进行富集分析,GO分析结果显示,显著性位列前5的生物过程包括转录共调节因子活性(GO:0003712)、细胞黏附分子结合(GO:0050839)、近端启动子序列特异性DNA结合(GO:0000987)、RNA聚合酶Ⅱ近端启动子序列特异性DNA结合(GO:0000978)、DNA结合转录激活剂活性/RNA聚合酶Ⅱ特异性(GO:0001228);而KEGG富集分析结果显示,差异表达基因主要富集在磷脂酰肌醇3-激酶(phosphatidylinositide 3-kinase,PI3K)-蛋白激酶B(protein kinase B,Akt)信号通路(hsa04151)、EB病毒感染(hsa04010)、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路(hsa05169)、人乳头瘤病毒感染(hsa05165)、人T细胞白血病病毒1感染(hsa05166)等信号通路。
2.3 WGCNA分析结果 通过WGCNA分析共获得4个模块(见图2A),其中绿松石模块共获得357个基因,蓝色模块共获得107个基因,棕色模块共获得91个基因,黄色模块共获得77个基因。由于绿松石模块包含的基因数最多,同时绿松石模块与蓝色和棕色模块有较高的相关性(见图2B),因此推测绿松石模块的基因可能在RA的发病机制中具有重要作用。KEGG富集分析结果显示,绿松石模块基因显著富集在趋化因子信号通路、细胞因子与细胞因子受体的相互作用、原发性免疫缺陷、T细胞受体信号传导途径、核因子κB信号通路、肿瘤坏死因子(tumor necrosis factor,TNF)信号通路、B细胞受体信号传导途径、破骨细胞分化、自然杀伤(natural killer,NK)细胞介导的细胞毒性等通路,见图2C;蓝色模块基因主要富集在光转导、细胞因子与细胞因子受体的相互作用、TNF信号通路、用于IgA产生的肠道免疫网络等信号通路;棕色模块基因主要富集在肺结核、FcγR介导的吞噬作用、RA、金黄色葡萄球菌感染等信号通路;黄色模块基因主要富集在过氧化物酶体增殖剂激活受体(peroxisome proliferator-activated receptor,PPAR)信号通路、AMP依赖的蛋白激酶(AMP-activated protein kinase,AMPK)信号通路、脂肪细胞中脂解的调控、药物代谢-细胞色素P450、脂肪细胞因子信号通路、酪氨酸代谢、脂肪酸降解等信号通路。
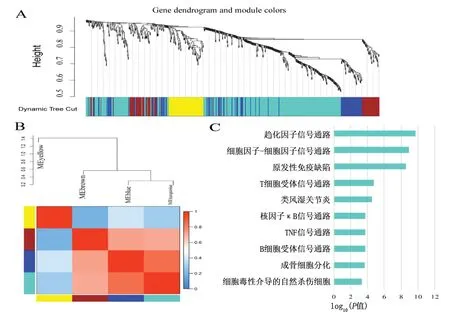
图2 WGCNA分析结果
2.4 免疫细胞浸润分析结果 通过比较正常滑膜组织和RA组织之间的免疫细胞浸润比例,发现RA滑膜组织中细胞毒性T细胞、1型调节性T细胞(type 1 regulatory T cell,Treg1)、B淋巴细胞、CD8+T淋巴细胞、中央记忆型T细胞浸润比例增多,而诱导调节性T细胞 (inducible regulatory T cell,iTreg)、Th17细胞、滤泡辅助性T细胞(follicular helper T cell,Tfh)、NK细胞、NK T淋巴细胞、中性粒细胞、CD4+T淋巴细胞浸润的比例减少,见图3。
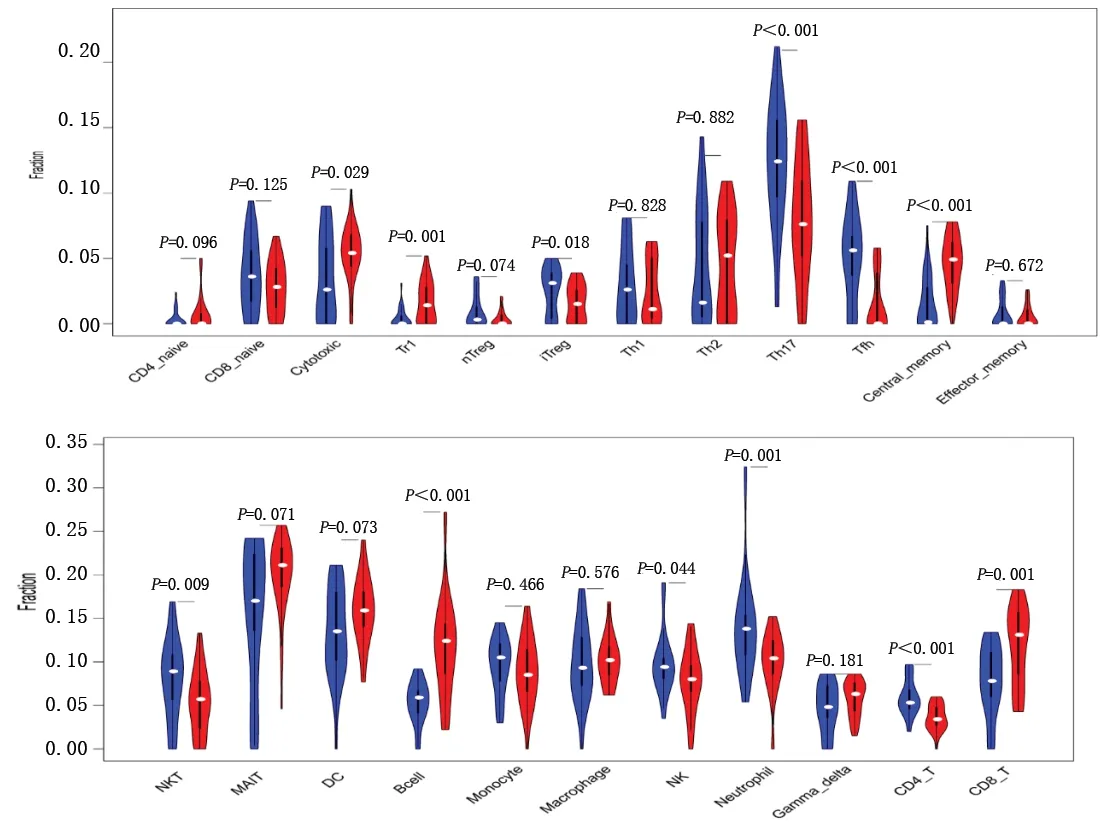
图3 免疫细胞浸润分析的小提琴图
2.5 免疫相关基因与免疫细胞的相关性研究 在RA差异表达基因中,共筛选到免疫相关基因234个,这234个基因共涉及52条通路。其中有2条与B淋巴细胞相关的信号通路,为B细胞受体信号通路和调节B细胞调节相关免疫,共涉及84个免疫相关基因;将这84个免疫相关基因进行PPI分析后,利用Cytohubb插件筛选关键基因(图4A),其中前5个最关键的基因为CR2、PAX5、CCL19、UBASH3A、TRAT1。进一步将这5个基因的表达量与免疫细胞浸润分析得到的B 淋巴细胞矩阵进行相关性分析,结果显示CR2、PAX5、CCL19、UBASH3A、TRAT1的表达量与B淋巴细胞矩阵呈正相关(r=0.533、0.750、0.669、0.769、0.741,P=0.001、<0.001、<0.001、<0.001、<0.001),见图5。
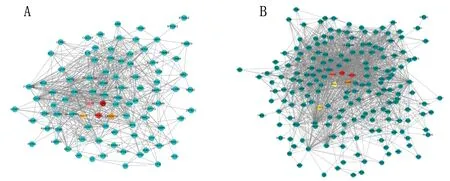
图4 免疫相关基因的PPI图
此外,共得到13条与T淋巴细胞相关的信号通路,包括T细胞的激活、T细胞的趋化等信号通路,涉及202个免疫相关基因,按照前述方法获得关键基因(图4B),其中前5个最关键的基因为CCR7、CXCR3、ITGAX、GZMB、PRF1。进一步将这5个基因的表达量与免疫细胞浸润分析得到的T淋巴细胞矩阵进行相关性分析,结果显示CXCR3、CCR7、PRF1、GZMB基因的表达量与CD8+T淋巴细胞矩阵呈正相关(r=0.571、0.853、0.746、0.596,均P<0.001),CXCR3、CCR7、PRF1基因的表达量与Treg1细胞矩阵呈正相关(r=0.432、0.374、0.263,P=0.010、0.027、0.045),CXCR3、CCR7、PRF1、GZMB基因的表达量与中央记忆型T细胞矩阵呈正相关(r=0.458、0.612、0.676、0.364,P=0.006、<0.001、<0.001、<0.001),CCR7、GZMB、PRF1基因的表达量与细胞毒性T细胞矩阵呈正相关(r=0.462、0.688、0.651,P=0.005、<0.001、<0.001),CCR7、GZMB基因的表达量与iTreg细胞矩阵呈负相关(r=-0.726、-0.491,P<0.001、0.003),见图5。
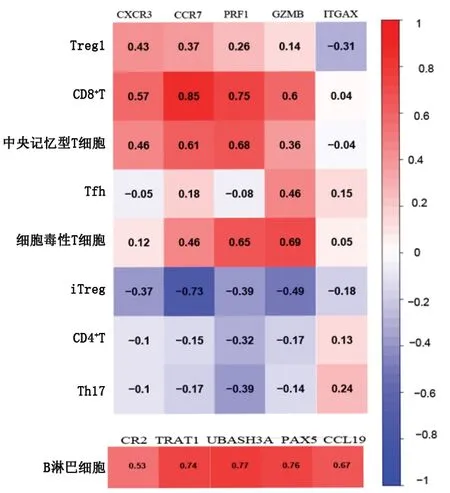
图5 关键免疫相关基因与T、B淋巴细胞的相关性分析
2.6 关键免疫相关基因在RA免疫细胞中表达的验证 单细胞测序数据集GSE141105共包含4 310个细胞,经注释后,共获得正常组B淋巴细胞136个和RA组B淋巴细胞1 165个,以及正常组CD4+T淋巴细胞81个和RA组CD4+T淋巴细胞369个。结果显示,RA组和正常组CD4+T淋巴细胞中仅ITGAX基因的表达差异存在统计学意义(均P<0.05),RA组和正常组B淋巴细胞中CR2、PAX5、UBASH3A基因的表达差异存在统计学意义(均P<0.05),见表2和表3。
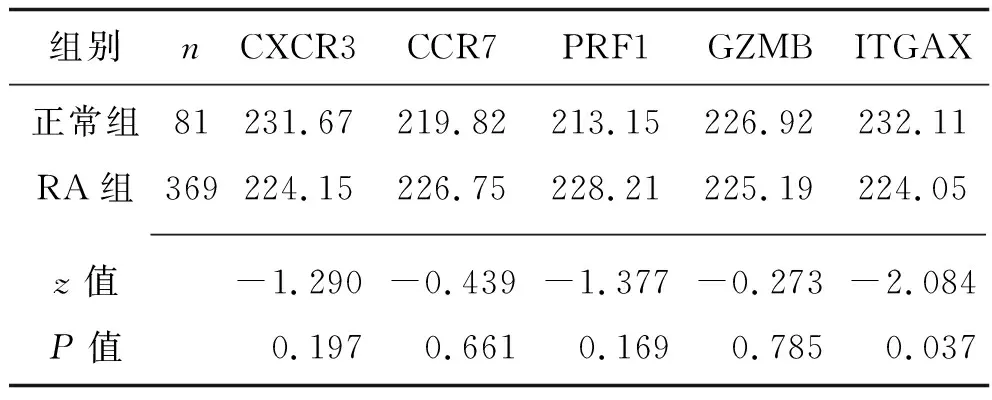
表2 正常组与RA组CD4+T细胞中关键基因表达量的比较

表3 正常组与RA组B淋巴细胞中关键基因表达量的比较
3 讨 论
本研究通过多芯片基因数据,共得到632个差异表达基因,其中最显著的10个差异表达基因分别为:PSMB9、VOPP1、GABARAPL1、SEL1L3、PLCG2、CXCL13、IGHM、IGKC、ADAMDEC1、MREG。其中,VOPP1通过促进核因子κB1的核易位来增加其转录活性[6],而核因子κB1与免疫系统密切相关;CXCL13是B淋巴细胞趋化因子;IGHM是B细胞的抗原识别分子;IGKC是一种蛋白质编码基因,与IGKC有关的疾病包括免疫球蛋白κ轻链缺乏症和浆细胞瘤;ADAMDEC1编码的基因属于整合素金属蛋白酶家族的分泌蛋白,树突状细胞成熟过程中其表达上调,该蛋白可能在树突状细胞功能及其与生发中心T细胞的相互作用中起重要作用。此外,我们还发现多个差异表达基因也与免疫相关,如TLR7、TNFSF11。其中,TLR7受体是天然免疫的一种模式识别受体,能够识别病原相关模式分子从而引发先天性免疫反应并启动获得性免疫反应。目前已知TLR7激动剂能够使初始B细胞增殖及活化,有研究表明滑膜组织释放的微小RNA-21能介导TLR7激活,导致膝骨关节炎疼痛,而TLR7拮抗剂还对膝盖骨性关节炎疼痛具有持久的镇痛作用[7]。TNFSF11是RANK的配体,在骨的分化中起重要作用,能通过激活破骨细胞前体细胞中的多个信号通路来诱导破骨细胞形成。同时,TNFSF11还参与免疫反应,能增强树突状细胞刺激幼稚T细胞增殖的能力,其可能是T细胞与树突状细胞之间相互作用的重要调节剂,并且可能在T细胞依赖性免疫应答的调节中发挥作用[8]。
我们进一步通过WGCNA分析将RA的差异表达基因划分为不同的模块。最为重要的绿松石模块富集在免疫反应和破骨细胞分化信号通路,说明免疫与骨分化紊乱可能是RA最重要的发病机制。而在免疫细胞浸润分析中我们也发现,滑膜组织中伴多种免疫细胞浸润,其中B淋巴细胞胞浸润比例增多。B淋巴细胞在RA发病中的作用已经被广泛证明。RA患者的滑膜组织中存在异位生发中心,B淋巴细胞可在滑膜组织中被激活;活化的B淋巴细胞参与抗原驱动的特异性免疫反应,可以分化成为高亲和力抗体的浆细胞;大多数RA患者血清中均可检测到自身抗体,人软骨糖蛋白39、Ⅱ型胶原蛋白、聚合素等均可能是体内的自身抗原,且60%~80%的RA患者中的自身抗体具有高亲和力[9];抗原与抗体形成免疫复合物,可以进一步激活补体,从而损伤关节滑膜组织等。此外,滑膜组织中的B淋巴细胞激活因子、TNF家族、CXCL13、CXCL1是影响滑膜组织中B细胞激活、分化和趋化的重要细胞因子[10]。本研究中,RA滑膜组织中差异表达基因TNFSF11、CCR5、CXCR3、CXCL1表达上调,提示这些B细胞相关细胞因子也促进了RA的发病。
本研究结果还显示,RA组和正常组B淋巴细胞中CR2、PAX5、UBASH3A基因的表达水平差异有统计学意义(均P<0.05)。CR2主要表达于人成熟B淋巴细胞的表面,在CR2-/-的小鼠腹膜中B淋巴细胞显著减少,且此类小鼠对T依赖性抗原的体液反应存在严重缺陷,其血清抗体滴度降低以及脾滤泡中生发中心的数量和大小减少,而该缺陷是由于B淋巴细胞本身引起的[11]。PAX5是早期B淋巴细胞分化的重要转录因子,该基因的靶向突变可在前体B淋巴细胞阶段阻止B细胞淋巴发育,且其还能通过抑制非B系基因以维持B淋巴细胞自稳[12]。PAX5转录可促进E2A缺陷型小鼠的前体 B淋巴细胞发育[13]。UBASH3A是编码T细胞泛素配体(TULA)家族的成员之一。有研究显示UBASH3A基因的多态性与系统性红斑狼疮等其他免疫性疾病相关[14],这提示UBASH3A基因对促进免疫性疾病的发生具有重要作用,但是具体机制仍需要进一步研究。此外,UBASH3A被认为是RA的易感基因。有学者对916例RA患者和2 266例健康者进行比较后发现,UBASH3A的遗传变异使汉族人更易发生RA,且UBASH3A基因rs1893592位点与RA的C反应蛋白水平和骨侵蚀疾病状态显著相关[15]。
此外,T淋巴细胞也广泛参与了RA的发病,T细胞在RA滑膜组织中处于慢性活化状态[16]。郑小娟等[17]分析了137例RA患者血清中Th的亚型,发现RA患者的Th总数增加,其中Th1/Th2、Th17/Treg高于健康人群。在本研究中,我们发现T淋巴细胞趋化因子受体CCR5及CXCR3在RA组织中表达上调,这提示T淋巴细胞浸润滑膜组织,可能是受到了趋化因子的影响。尽管有实验已经证明CD4+T淋巴细胞参与了RA的发病,RA患者外周血CD4+T淋巴细胞比例升高[18],但是本研究结果显示,RA组滑膜组织中CD4+T淋巴细胞浸润比例减少,而CD8+T淋巴细胞浸润比例增高。Cho等[19]也发现,在RA患者的滑膜组织中,产生IL-10的抑制性CD8+T淋巴细胞数量显著增加。而另外一项研究结果亦显示,RA患者滑膜组织中效应记忆性CD8+T淋巴细胞比例显著高于外周血标本,且表达CD25和CD69的CD8+T淋巴细胞比例明显高于外周血标本[20]。以上均提示CD8+T淋巴细胞在RA发病中发挥重要作用。
综上所述,免疫紊乱可能是RA的重要发病机制之一,RA患者滑膜组织中细胞毒性T细胞、Treg1细胞、B淋巴细胞、CD8+T淋巴细胞浸润数量增多,其中CR2、PAX5、UBASH3A基因表达与B淋巴细胞显著相关,这些免疫细胞和免疫相关基因可能在RA的发生中发挥重要作用。但是本研究验证差异基因在免疫细胞中的表达时,由于GSE141105数据集滑膜组织中所获得的细胞量较少,且单细胞测序数据包含较多差异表达量为0的数据,故本研究选择在RA小鼠外周血单细胞集中进行验证,但由于RA主要病变位于滑膜,故今后仍需要在滑膜组织中进一步验证;此外,本研究仅采用生物信息学的方法进行分析,今后仍需要进行实验研究验证所得结论。
