脊髓小脑性共济失调6型一家系
2021-09-22杨云鹏刘佳王鲁宁
杨云鹏 刘佳 王鲁宁
先证者 女性,41岁。主因头晕、行走不稳、言语不清4年并进行性加重2年,于2016年5月21日入院。患者4年前无明显诱因出现头晕、步态不稳、步基增宽等症状,但尚能独立行走,同时伴有言语不清,语调、语速改变,近2年来不仅新增饮水呛咳,而且上述症状呈进行性加重,多次于外院就诊,均以脑供血不足而予血塞通、天麻素等药物治疗,但病情仍不断进展,为求进一步诊断与治疗,遂至我院就诊,门诊以头晕原因待查收入神经内科。自患病以来,精神稍差,睡眠可,饮食正常,大小便正常,体重无明显变化。
既往史、个人史及家族史 既往身体健康,否认特殊慢性疾病病史,父母非近亲婚配。其父现年73岁,身体健康,其母71岁,于33岁出现行走不稳症状;另有兄弟姊妹5人,除三兄30岁时死于车祸,长兄目前尚未发病,其余3人中二姐现年51岁、四兄43岁、六弟39岁,均于36岁时出现与先证者相类似的症状;先证者母亲及二姐因下肢运动障碍失去行走能力。
诊断及治疗经过 入院后体格检查:神志清楚,对答切题,呈爆破性语言,双眼各向活动正常,可见粗大的水平眼震。行走时步基增宽,四肢肌力正常,肌张力减低;双侧肱二头肌反射、肱三头肌反射、桡骨膜反射、跟-膝-腱反射存在。双侧指鼻试验欠稳准,双侧快复轮替试验笨拙,跟-膝-胫试验欠稳准,直线行走不能,Romberg征阳性,肌回缩实验(反击征)阳性。双上肢意向性震颤。双侧掌颌反射阳性,右侧Babinsik征阳性,左侧Babinsik征、双侧Chaddock征阴性。深浅感觉正常,脑膜刺激征阴性。洼田饮水试验2级。神经功能量表检查:经征得先证者及其母、二姐、四兄、六弟知情同意,均于我院接受国际协作共济失调评价量表(ICARS)检查,检查结果显示:总评分分别为44、84、64、26和25,其中姿势和步态障碍静态评分为18、32、26、9和8,动态肢体协调评分为21、44、30、13和13,语言障碍评分为3、4、4、2和2,眼球运动障碍评分为2、4、4、2和2(表1)。先证者实验室各项检查指标均无明显异常。心脏、颈部血管及腹部彩超检查无异常发现。影像学检查:先证者T2WI平扫显示小脑萎缩,以小脑蚓部明显,脑干轻度萎缩(图1a)。根据家族史分别对先证者之母、二姐、四兄、六弟进行MRI检查,其四兄及六弟既往保存有完整的MRI检查资料,亦呈小脑萎缩表现(图1b,1c)。结合先证者家族史、临床特征、影像学检查结果,考虑遗传性共济失调疾病,且符合常染色体显性遗传性疾病遗传特点。在征得先证者及其子女,家族中其父母、长兄及其子、二姐及其女、三兄之子女、四兄及其子、六弟及其子女共16人知情同意,采集静脉血5 ml,采用聚合酶链反应(PCR)联合毛细管电泳(CE)法对临床常见脊髓小脑性共济失调(SCA)1、2、3、6、7、12、17型,以及齿状核红核苍白球路易体萎缩(DRPLA)等常染色体显性遗传性共济失调(ADCA)亚型进行基因检测(北京金准基因科技有限责任公司),结果显示:先证者(Ⅱ9)CACNA1A基因的1个等位基因CAG拷贝数超出正常范围,为23次;家族中先证者之母(Ⅰ2)、二姐(Ⅱ3)、四兄(Ⅱ7)、先证者之女(Ⅲ9)、六弟(Ⅱ11)及其子(Ⅲ)11共6人存在CACNA1A基因CAG重复扩增,扩增次数均超出正常范围,为23次,符合SCA6型致病特点,该家系明确为SCA6型家系(图2)。治疗原则以营养神经、促进神经细胞代谢为主,予以胞二磷胆碱0.50 g/d静脉滴注、辅酶Q10口服10 mg/次(3次/d)、丁苯肽口服200 mg/次(3次/d)。患者共住院10天,出院时头晕、行走不稳、构音障碍等症状无明显改善,出院后继续口服胞二磷胆碱200 mg/次(3次/d)和银杏叶提取物40 mg/次(3次/d)。3个月复诊时,症状与体征仍如前。

图1 头部横断面T2WI显示小脑萎缩,以小脑蚓部明显;脑干轻度萎缩(箭头所示) 1a 先证者 1b 先证者四兄 1c先证者六弟Figure 1 Brain imaging showed cerebellar atrophy on axial T2WI,especially in vermis and mild atrophy in brainstem(arrows indicate) The proband(Panel 1a).The fourth brother of the proband(Panel 1b).The sixth brother of the proband(Panel 1c).
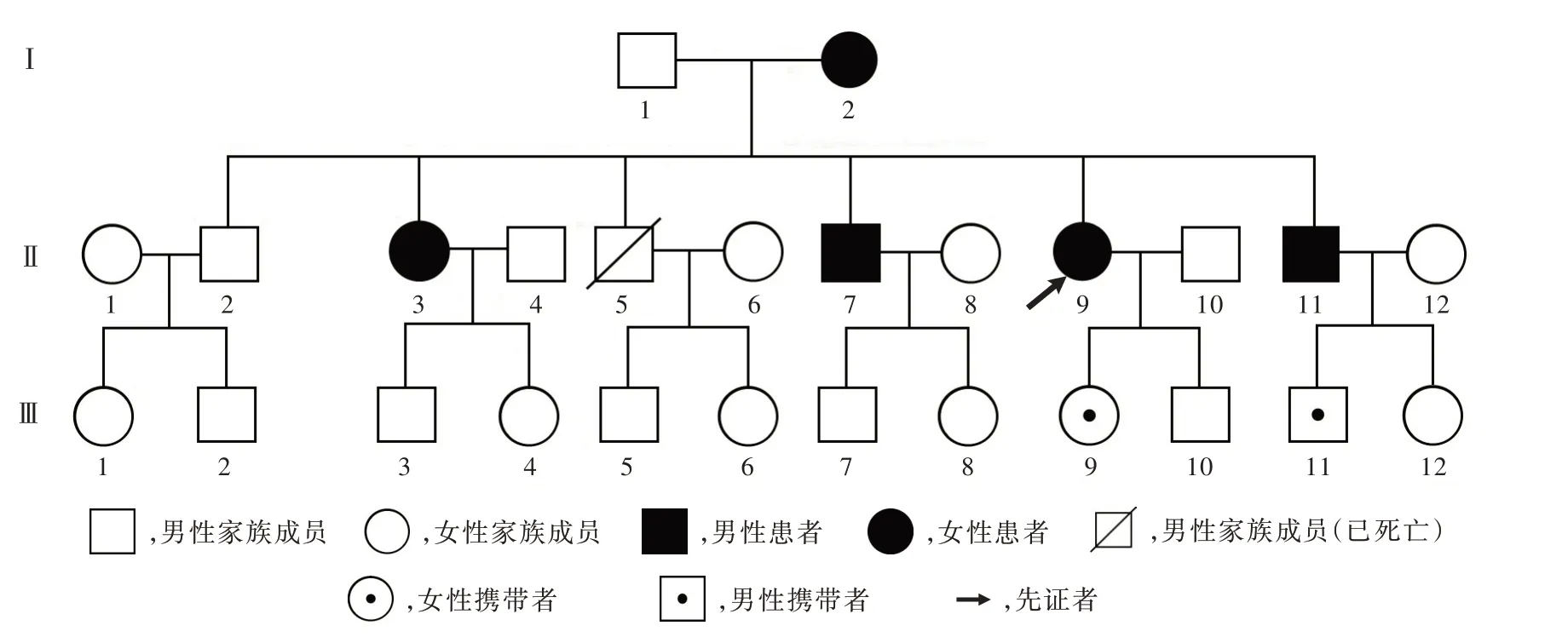
图2 SCA6型家系图Figur e 2 The SCA6 pedigree of three generations of the patient's family.
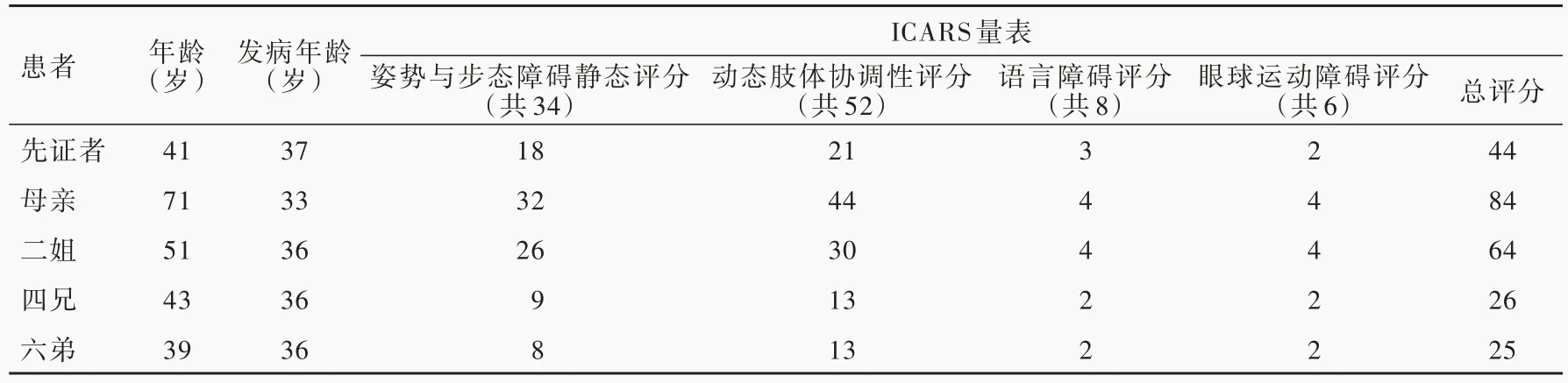
表1 SCA6型家系ICARS量表检查结果Table 1.Results of ICARSscale of SCA6 pedigree
讨 论
遗传性共济失调是一大类具有高度临床和遗传异质性、病死率和病残率均较高的遗传性神经系统退行性疾病,占神经系统遗传性疾病的10%~15%[1]。遗传性共济失调多于成年期(>30岁)发病,以小脑性共济失调为主要特征,表现为平衡障碍、进行性肢体协调运动障碍、步态不稳、构音障碍、眼球运动障碍等,同时可伴有舞蹈症、帕金森样症状、肌张力障碍等锥体外系症状[2],以及锥体系、视觉、听觉、脊髓、周围神经损害,亦可伴大脑皮质功能损害如认知功能障碍和(或)精神行为异常等。本文家系三代共16人接受基因检测,其中先证者母亲、二姐、四兄、六弟及其子、先证者及其女共7例存在CACNA1A基因变异,其中先证者及其母、二姐、四兄、六弟共5例已经出现不同程度的小脑性共济失调症状,发病年龄为33~37岁,该家系第三代2例CACNA1A基因变异携带者年龄<18岁(分别为9岁和12岁),目前尚无临床症状,符合成年期发病之特点。与其他SCA类型相比,SCA6型发病较晚。本文家系中5例患者均接受ICARS量表检查,该量表是半定量化的神经功能评价量表,可以描述和定量评价典型的小脑性共济失调症状,包括姿势和步态障碍评分、动态功能评分、语言障碍、眼球运动障碍评分,总分为100,评分越高,提示协调功能障碍越严重,本文家系中以先证者母亲ICARS量表评分最高,其次为其二姐、先证者、四兄、六弟,5例患者的评分均与其病程相关。该家系患者语言障碍和眼球运动障碍相对步态障碍、肢体协调症状要轻,且均以步态异常为首发症状并进行性加重,其中年龄较大的先证者之母及其二姐目前已不能独立行走,提示SCA6型病例大多以步态共济失调发病且可导致严重残疾。研究表明,SCA6型患者在临床症状出现之前即已存在选择性步态障碍,其步态障碍对疾病进展具有较高的敏感性[3],且平衡障碍与其疾病严重程度呈正相关,是SCA6型临床综合征进展的特征性表现[4]。本文先证者洼田饮水试验为2级,家系中所有患者均无明显吞咽困难、饮水呛咳主诉。来自日本的一项研究表明,SCA6型患者所表现的吞咽困难通常比SCA3型轻,该研究经对14例SCA6型患者进行反复的荧光透视检查(videofluoroscopic examinations)发现,其吞咽困难总体进展十分缓慢[5]。国内文献曾报道SCA6型两家系临床表现特点:(1)遗传早现现象。(2)于中年期发病,病程进展缓慢,症状相对较轻。(3)躯干和(或)肢体共济失调。(4)慢眼活动。(5)假性延髓麻痹。(6)眼震。(7)意向性震颤。(8)腱反射减弱或活跃。(9)病理征阴性。(10)头部MRI显示小脑萎缩,而脑干基本正常[6]。本文家系临床特点与上述基本相似,但也存在一些差异,本文家系未见明显的遗传早现现象,头部MRI可见小脑萎缩,以小脑蚓部明显,脑干轻度萎缩;且病理征呈阳性。国外也有文献报道,SCA6型患者小脑蚓部萎缩较半球更为严重,且可累及脑干,类似于橄榄脑桥小脑萎缩(OPCA)改变[7]。本文首次报告我国云南省大理白族自治州SCA6型家系且病例数较多,共16人行SCA1、2、3、6、7、12、17,以及DRPLA亚型相关基因检测,其中7例CACNA1A基因存在异常扩增,为大理地区遗传性共济失调流行病学研究奠定了基础。
SCA6型目前尚无有效的治疗药物,临床上仍以对症和支持治疗为主,主要目的是减轻症状、延缓病情进展,改善日常生活自理能力。主要治疗药物包括神经保护剂辅酶Q10、艾地苯醌、利鲁唑等,虽然这些药物的神经保护效果尚有待证实,但有研究显示,利鲁唑具有改善遗传性共济失调患者运动障碍症状的作用[8-9]。对症治疗药物包括丁螺环酮、坦度螺酮,可改善患者共济失调症状;左旋多巴及其复合制剂、苯海索、金刚烷胺等则用于改善锥体外系症状。非药物治疗主要包括神经康复、经颅磁刺激、心理治疗等。此外,基因治疗和干细胞移植亦具有广阔的应用前景,尚待进一步研发。
利益冲突无
