特发性震颤相关基因研究进展
2021-09-22万雅兰王朝霞
万雅兰 王朝霞
特发性震颤(ET)亦称为原发性震颤,是临床常见的运动障碍性疾病,患病率为0.9%,并且随年龄的增长而明显升高,65岁以上人群患病率为4.6%、95岁以上人群可达20%[1]。特发性震颤主要表现为姿势性震颤和动作性震颤,最常累及手部及上肢,亦可累及下肢、头面部、躯干及发音。发病机制迄今尚未阐明,20世纪70年代提出的“下橄榄核模型”认为,下橄榄核中起搏神经元过度激活导致橄榄-小脑信号输出异常,引起震颤[2];“小脑退化模型”成为近年研究热点,该模型基于特发性震颤患者小脑存在浦肯野细胞轴突和树突改变及浦肯野细胞移位和丢失、篮状细胞轴突改变、攀缘纤维与浦肯野细胞连接异常分布,以及齿状核γ-氨基丁酸受体(GABAR)改变,进而提出小脑病变是导致特发性震颤的关键原因[3]。基于脑电图(EEG)、脑磁图(MEG)和fMRI的神经网络研究显示,特发性震颤患者皮质-橄榄-小脑-丘脑环路活动增强,此环路将现有的特发性震颤相关理论较好地进行概括,且为脑深部电刺激术(DBS)以及周围电刺激术提供理论基础[4](图1)。流行病学调查发现,约50%的特发性震颤患者有家族史,≥80%的早发型(40岁前发病)患者存在1个及以上的一级亲属患病[5],揭示遗传因素在特发性震颤的发病过程中起重要作用。既往研究多认为特发性震颤高度遗传且为单基因常染色体显性遗传性疾病,至65岁时可完全外显[6]。但近5年的观点认为,特发性震颤是一种基因因素与环境因素共同作用的疾病,且存在基因不完全外显的情况[7],相关表观遗传学研究较少。2019年,Paul等[8]对12例特发性震颤患者进行尸检,发现小脑甲基化改变可能与特发性震颤的发病有关。而其他表观遗传学机制,如组蛋白修饰、微小RNA(miRNA)表达等暂未见诸报道。尽管目前仅有特发性震颤常染色体显性遗传模式的报道,但仍有学者认为其可能存在双基因或多基因致病[7]。
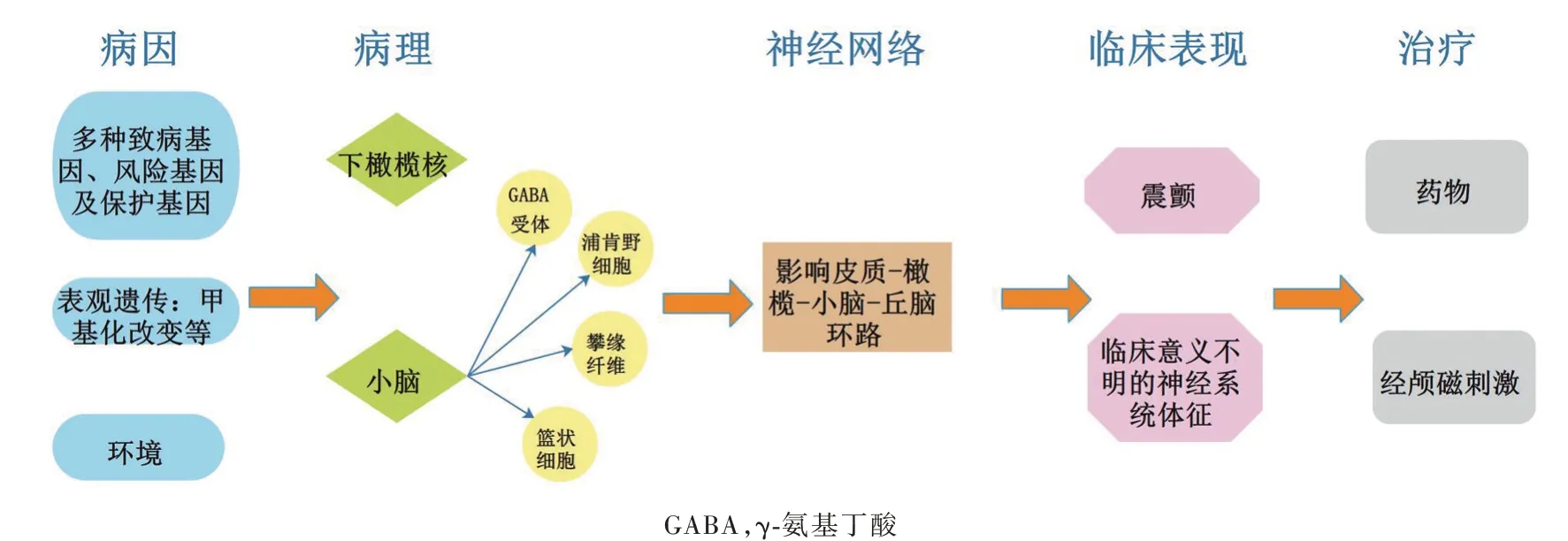
图1 特发性震颤的疾病模式:基因、表观遗传及环境因素共同作用,导致下橄榄核及小脑病变,进而影响皮质-橄榄-小脑-丘脑环路,最终导致震颤,伴或不伴临床意义不明的神经系统体征Figure 1 Disease model of essential tremor:genetic,epigenetic and environmental factors work together to cause lesions in the inferior olive nucleus and cerebellum,which in turn affect the cortical-olive-cerebellum-thalamus circuits,and ultimately lead to tremor,with or without neurological signs of uncertain clinical significance.
20世纪90年代以来,陆续有学者通过特发性震颤 家 系 连 锁 分 析(linkage analysis)确 定ETM1、ETM2、ETM3共3个基因座;以及通过全外显子组测序(WES)在个别患者或家系中发现FUS、TENM4、SCN4A、SCN11A、KCNS2、HTRA2、HAPLN4、SORT1、NOS3基因变异,全基因组测序(WGS)发现CACNA1G基因变异,全基因组关联分析(GWAS)发现LINGO1、LINGO2、SLC1A2、STK32B、PPARGC1A、CTNNA3等基因单核苷酸多态性(SNP)位点与特发性震颤相关[5]。2020年,唐北沙教授团队采用长读长高通量测序(LRS)发现,特发性震颤患者存在NOTH2NLC基因5’非翻译区(5'UTR)鸟嘌呤-鸟嘌呤-胞嘧啶(GGC)重复扩展突变[9](图2),未来有望通过第三代测序技术发现其他相关基因。

图2 特发性震颤致病基因以及风险或保护基因研究的里程碑Figure 2 Milestones in the study of essential tremor pathogenic genes and risk/protective genes.
目前已发现的特发性震颤致病基因有10余个,风险或保护基因也有10余个[5],这些基因涉及的细胞生物学功能包括调控细胞膜离子通道、小胶质细胞功能及髓鞘形成、细胞凋亡、γ-氨基丁酸(GABA)能系统、线粒体功能、细胞核与细胞质之间物质转运等(图3)。其中一些基因如DRD3、HS1BP3、TENM4、CACNA1G、HAPLN4,在皮质-橄榄-小脑-丘脑环路中呈高表达,其基因变异可能通过直接影响环路而引起震颤;另一些基因如FUS、USP46、LINGO1,在神经系统呈广泛表达,但这些基因变异导致的特发性震颤却仅表现为震颤,推测可能是由于皮质-橄榄-小脑-丘脑环路中神经元较其他脑区有一定特殊性,尚待更多基础研究加以证实;此外,还有一些基因例如STK32B、PPARGC1A、CTNNA3,在神经系统表达较少,主要表达于其他组织器官如心脏、肾脏,其变异致震颤的作用机制鲜有报道(表1,2)[9-24]。

表1 特发性震颤相关致病基因Table 1.Essential tremor related pathogenic genes
一、特发性震颤相关致病基因
1.ETM1、ETM2、ETM3基因座 ETM1、ETM2和ETM3是通过连锁分析定位的3个与特发性震颤相关的基因座。2006年,Jeanneteau等[25]在23个法国特发性震颤家系中发现位于ETM1基因座的DRD3基因p.Ser9Gly变异。DRD3基因在基底节区呈高表达,在小脑浦肯野细胞也有表达。DRD3蛋白是G蛋白耦联受体(GPCR)多巴胺D2受体(D2R)家族成员,通过抑制腺苷酸环化酶(AC)而抑制c AMP/蛋白激酶A(PKA)信号转导通路,而p.Ser9Gly变异可加强此抑制作用,从而导致特发性震颤[26]。2005年,Higgins等[27]在2个美国特发性震颤家系中发现位于ETM2基因座的HS1BP3基因p.Ala265Gly变异。HS1-BP3蛋白在运动神经元和浦肯野细胞呈高表达,通过与磷脂酸结合以减少细胞自噬体形成,促使细胞凋亡,从而导致特发性震颤[28]。ETM3基因座定位于6p23,理论上应该存在致病基因,但对该区域的15个基因进行测序并未确定特发性震颤相关致病基因[17],尚待进一步研究证实。
2.FUS基因FUS基因位于ETM4基因座,对基因修复、基因转录、RNA加工、细胞增殖均有调节作用[29]。2012年,Merner等[13]报告一特发性震颤家系存在FUS基因p.Gln290*变异。该变异可阻碍核蛋白输出,也可通过损害GABA信号转导通路导致运动障碍。
3.TENM4基因TENM4基因位于ETM5基因座。2015年,Hor等[14]报告3个西班牙特发性震颤家系中存在TENM4基因p.Ala1442Thr变异,但该变异在后续其他研究中未能得到验证[30]。TENM4蛋白主要表达于小脑白质,可调节少突胶质细胞分化,并在小直径轴突髓鞘形成过程中发挥重要作用。p.Ala1442Thr变异通过造成小脑白质损害,继而引起特发性震颤[31]。
4.电压门控离子通道相关致病基因 钠、钾、钙离子通道相关基因与特发性震颤之间的关系于2015-2019年陆续见诸报道[15-18],这些基因多与调节神经元兴奋性有关,但导致特发性震颤的具体机制尚缺乏研究证实。有研究显示,编码钠离子通道α亚基Nav1.4的SCN4A基因p.Gly1537Ser变异和编码Nav1.9的SCN11A基因p.Arg225Cys变异均与特发性震颤有关[32]。SCN4A基因高表达于肌肉以及
神经系统,通过降低丘脑神经元动作电位阈值以引起震颤。SCN4A基因变异影响钠离子通道的离子选择性,降低重复动作电位激发幅度,并影响钾离子和氨基酸的转运,从而影响丘脑相关的连接通路,引起特发性震颤[33]。SCN11A基因变异通过延长阈下刺激的去极化,调节静息神经元的易兴奋性,增强神经系统兴奋性,继而引起特发性震颤[5]。2019年,Hosoi等[34]在特发性震颤小鼠模型中发现,编码Nav1.6的SCN8A基因与特发性震颤具有相关性,但迄今尚未在特发性震颤患者中发现这一变异。2016年,Liu等[17]报告编码钾离子通道α亚基Kv9.2的KCNS2基因p.Asp379Glu变异与特发性震颤有关。KCNS2基因高表达于小脑浦肯野细胞和颗粒细胞,对调节高频率突触刺激具有重要作用。钾离子通道Kv3家族也在小脑浦肯野细胞表达[35],但其与特发性震颤的关系尚未见诸文献报道。2019年,Odgerel等[18]在一特发性震颤家系中发现CACNA1G基因p.Arg456Gln变异。CACNA1G基因高表达于小脑浦肯野细胞和小脑深部核团[36],主要作用是产生短脉冲电信号,作用于非橄榄小脑束起源的运动通路,导致特发性震颤,但其具体机制尚待进一步研究[37]。
5.线粒体功能及细胞凋亡相关致病基因Unal Gulsuner等[19]于2014年在一土耳其特发性震颤大家系中发现HTRA2基因变异。当神经细胞受到凋亡性刺激时,HTRA2蛋白自线粒体膜间释放入细胞质,参与启动细胞凋亡[38]。HTRA2基因p.Gly399Ser变异可导致线粒体功能紊乱、形态异常,降低蛋白激酶活性,过表达此变异的细胞在应激时更易死亡。HTRA2基因p.Gly399Ser变异为功能缺失变异,其导致特发性震颤可能与纹状体神经元缺失有关[19],但这一观点尚缺乏相关研究证实。
6.GABA信号转导通路相关致病基因 震颤小鼠模型显示,注射γ-氨基丁酸A型受体(GABAAR)激动剂可减轻小鼠震颤症状[39],提示GABA信号转导通路表达下调与特发性震颤的发病有关。SORT1[20]、HAPLN4[17]、USP46[17]这3个GABA信号转导通路基因变异可能与特发性震颤的发病相关。2015年报道的一西班牙特发性震颤家系中存在SORT1基因p.Gly171Ala变异[20]。SORT1基因编码的Sortlin蛋白在中枢神经系统广泛表达,Sortlin蛋白不仅通过调节蛋白转运和信号转导调节神经元功能,还通过与神经生长因子前体结合调控神经元存亡。SORT1基因p.Gly171Ala变异可导致Sortlin蛋白减少,而与Sortlin结合的P75NTR蛋白增多,后者增多见于GABA信号转导通路受损[5]。2016年的一项针对37个早发型特发性震颤家系的研究发现HAPLN4基因p.Gly350Arg变异及USP46基因p.Ala133Val变异[17]。推测上述3个基因变异均通过下调GABA信号转导通路而导致特发性震颤,但二者之间的相关性尚缺乏多中心大样本研究验证。
7.一氧化氮信号转导相关基因 一氧化氮在神经系统中参与介导神经元的生存、突触可塑性、血管平滑肌细胞松弛和血管内皮细胞渗透性[40]。2016年,Liu等[17]对37个早发型特发性震颤家系进行研究并报告NOS3基因p.Gly16Ser及p.Pro55Leu变异。NOS3基因高表达于中枢神经系统(尤其是小脑)神经元和血管内皮细胞,编码一氧化氮合酶(NOS),其导致特发性震颤的具体机制尚待进一步研究[40]。
8.NOTCH2NLC基因三核苷酸拷贝数扩增突变 2020年,我国学者Sun等[9]通过LRS测序对197个特发性震颤家系进行研究,发现NOTCH2NLC基因5'UTR GGC拷贝数扩增,约5.58%的患者携带该变异。NOTCH2NLC基因高表达于神经系统放射状胶质细胞,主要作用是增强Notch信号转导通路的表达,从而对大脑发育、神经元增殖和分化发挥至关重要的作用。但是位于NOTCH2NLC基因5'UTR的动态变异可能并非通过导致编码蛋白异常表达而致病,其作用机制更加复杂,目前有RNA毒性作用、重复相关非ATG翻译等假说[41],尚待进一步研究验证。
二、特发性震颤相关风险和保护基因
1.特发性震颤相关风险基因 (1)LINGO基因家族:2009年,Stefansson等[21]在452例冰岛特发性震颤患者中发现LINGO1基因rs9652490和rs11856808单核苷酸多态性位点。全基因组的单核苷酸多态性微阵列分析发现,LINGO1基因在特发性震颤患者中存在拷贝数扩增[42]。LINGO1基因广泛分布于不同脑区,通过与P75NTR或肿瘤坏死因子受体超家族成员19(TROY19)共同形成轴突生长抑制蛋白-66受体1(NgR1)复合物,参与神经元分化、少突胶质细胞成熟、抑制轴突生长、加速多巴胺能神经元变性死亡。病理学研究表明,特发性震颤患者LINGO1基因主要表达于小脑篮状细胞轴突内,在浦肯野细胞轴突起始部分形成刷状结构,从而影响浦肯野细胞功能[5]。此外,LINGO1蛋白作为钙离子激活的钾离子通道亚单位,可以引起钾离子通道的功能性敲除,导致特发性震颤及运动障碍[43]。2010-2011年,陆续有学者发现LINGO2基因rs1412229、rs7033345、rs10812774单核苷酸多态性位点[22]。LINGO2基因仅表达于中枢神经系统,关于其引起特发性震颤的研究甚少,有学者认为其与LINGO1基因功能相似[22]。(2)其他热点特发性震颤风险基因:2012年,Thier等[23]对990例特发性震颤患者和1537例健康对照者进行病例对照研究,发现SLC1A2基因rs3794087单核苷酸多态性位点。SLC1A2基因编码的谷氨酸再摄取转运蛋白兴奋性氨基酸转运体2(EAAT2)主要作用为将谷氨酸从突触间隙中移除,从而解除谷氨酸的神经毒性作用。SLC1A2基因表达于浦肯野细胞轴突起始部分,在特发性震颤患者小脑皮质表达明显减少[5]。2016年,Müller等[24]发 现 ,PPARGC1A基 因rs17590046及CTNNA3基因rs12764057、rs10822974、rs7903491单核苷酸多态性位点均可增加特发性震颤的患病风险,但并未在后续研究中得到证实[44]。PPARGC1A基因编码过氧化物酶增殖激活受体γ共受体1α(PGC-1α),通过与解耦联蛋白2及核呼吸因子相互作用调节线粒体功能及能量代谢,尸检研究显示,帕金森病患者黑质及苍白球PGC-1α异构体表达升高[45]。CTNNA3基因编码的α降钙素3在细胞-细胞黏附过程中发挥重要作用,但其导致特发性震颤的机制尚待进一步研究验证[44]。
2.特发性震颤相关保护基因 2016年,Müller等[24]对2807例欧洲特发性震颤患者及6441例健康对照者进行病例对照研究,发现STK32B基因rs10937625单核苷酸多态性位点是特发性震颤的保护因素,且这一结论在中国学者针对218例特发性震颤患者的GWAS研究中得以验证[46]。STK32B基因编码丝氨酸/苏氨酸激酶,但其功能尚不十分清楚。推测STK32B基因在特发性震颤患者小脑皮质的表达升高,而rs10937625单核苷酸多态性位点可以导致STK32B基因在小脑皮质的表达减少,从而成为特发性震颤的保护因素。
三、总结与展望
特发性震颤的遗传特征复杂,致病基因谱十分宽泛[47]。值得注意的是,目前已报道的致病基因在特发性震颤患者中的阳性检出率均很低,尚未发现高频率的致病基因,因此特发性震颤并非某一种单一致病基因的疾病,而是一组遗传异质性谱系疾病。此外,疾病易感基因和表观遗传修饰也与特发性震颤的发病有关。因此,特发性震颤的诊断应以病史、临床表现及体格检查为基础,对经济条件允许的患者可进行基因检测以辅助诊断与鉴别诊断,但不能过度依赖基因检测结果。在对基因检测结果进行分析时,应注意测序结果的判读,对检测到的变异应结合既往报道谨慎分析是否为致病性变异。总之,特发性震颤相关基因研究有助于临床医师对疾病的认识更加深入。未来研究方向为,通过新技术发现更多新的变异基因及位点、深入研究基因变异致病机制的同时,紧密结合临床,阐释不同变异位点与发病年龄、临床症状、药物反应的关系,从而为精准治疗提供线索。
利益冲突无
