眼咽型肌营养不良症一家系临床及分子生物学特征分析
2021-09-22黄铠李文武刘红仙孙浩李志宏褚嘉祐杨昭庆
黄铠 李文武 刘红仙 孙浩 李志宏 褚嘉祐 杨昭庆
眼咽型肌营养不良症(OPMD)是一种晚发型常染色体显性或隐性遗传性肌肉病,临床主要表现为成年发病的进行性眼睑下垂、吞咽困难和肢体无力等,较少伴神经系统病变[1]。其发病与PABPN1基因外显子1(GCG)异常重复扩增或(GCA)插入导致的PABPN1基因在肌细胞胞核内形成类淀粉丝样结构有关[2]。目前国内外均有报道,欧美等发达国家患病率较高,亚洲国家中以日本患病率最高,我国仅为个案报道[2],明确诊断依靠临床特点及PABPN1基因检测。男女均可发病,高峰发病年龄为40岁后,核心症状为眼睑下垂和吞咽困难,病情进展缓慢,首发症状通常为眼睑下垂,数年后可出现吞咽困难和构音障碍,饮水呛咳少见,亦有少数患者以吞咽困难为首发症状[3];实验室检查血清肌酸激酶(CK)水平正常或轻度升高[2];骨骼肌CT或MRI检查显示椎旁肌、腰大肌、背部深部肌群萎缩以及肢体远端和近端肌肉受累[2];肌肉组织活检可为鉴别诊断和基因诊断提供线索[4-6];绝大多数患者肌电图呈肌源性损害,少数可见动作电位时限增宽和波幅增高等神经损伤表现[7-8]。检测手段的增加及检测技术的进步可为疾病诊断提供更多依据。1998年,Brais等[4]的连锁分析显示,OPMD患者第14号染色体长臂(14q11.2)PABPN1基因外显子1由正 常 的(GCG)6(GCA)3(GCG)扩 增 为(GCG)8~13(GCA)3(GCG)。2011年 ,Robinson等[9]发 现 ,PABPN1基因外显子1存在点突变(35G>C)。GCA、GCT、GCC、GCG均 编 码 丙 氨 酸[10],因 此 以GCN代表上述四种中任意一种,正常等位基因(GCG)6即以(GCN)10替代,常染色体显性遗传性OPMD患者PABPN1基因通常为(GCN)12~17,而欧美和日本OPMD家系的基因变异类型以(GCN)13居多,约占40%,其次为(GCN)14和(GCN)15,分别占26%和21%,而(GCN)12、(GCN)16和(GCN)17较少见[4],其中 ,(GCN)13[11]和(GCN)15[12]在 我 国 均 有 报 道 。(GCN)17基因型患者较(GCN)12基因型发病年龄更早、病情更严重,尤以显性遗传性纯合子基因型症状最严重,纯合子(GCN)13的临床症状较杂合子(GCN)13和纯合子(GCN)11更严重[5-6]。常染色体隐性遗传性OPMD的PABPN1基因纯合子(GCN)17发病较晚且症状较轻,而(GCN)13变异和杂合子(GCN)11较纯合子(GCN)11的临床表型更严重[4]。本研究收集并分析一常染色体隐性遗传性OPMD家系的临床及分子生物学特点,进一步补充疾病的临床表型异质性。
对象与方法
一、研究对象
征得先证者及其亲属知情同意后,选择2019年12月在中国医学科学院北京协和医学院医学生物学研究所经基因检测明确诊断的OPMD患者3例及其直系亲属3名,分别为先证者(Ⅱ3)、先证者之大姐(Ⅱ1)、先证者二姐(Ⅱ2)、先证者四弟(Ⅱ4)、先证者之五妹(Ⅱ5)、先证者之六妹(Ⅱ6),均来自云南省楚雄彝族自治州的同一家系(图1)。其中,男性2例,女性4例;年龄50~65岁,平均58岁。
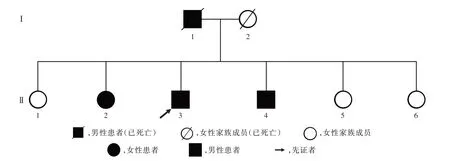
图1 OPMD患者家系图Figure 1 Family pedigree of patients with OPMD.
二、研究方法
1.临床资料采集 详细记录先证者一般情况(包括性别、年龄、首发症状、临床主要表现等)、既往史、个人史、家族史、体格检查以及各项辅助检查结果。
2.基因检测 采集6例家族成员外周静脉血各2 ml,置于乙二胺四乙酸(EDTA)抗凝管中,采用Axygen血液基因组DNA分离试剂盒[爱思进生物技术(杭州)有限公司]提取血液基因组DNA。根据GenBank数 据 库(https://www.ncbi.nlm.nih.gov/genbank/)获取PABPN1基因(NC_000014.9)外显子1全序列,采用Primer 5.0软件设计聚合酶链反应(PCR)引物,于昆明硕擎生物科技有限公司合成上游引物(F):5'-CGCAGTGCCCCGCCTTAGA-3'、下游引物(R):5'-ACAAGATGGCGCCGCCCCGGC-3'。以基因组DNA作为模板对目标基因序列行PCR扩增,PCR反应体系:基因组DNA(90 ng)4μl,10 mmol/L上游和下游引物各0.50μl,5 U/μl LA Taq 0.30μl,2×TSINGKE Master Mix(北京擎科生物科技有限公司)12.50μl,去离子水补足体系至25μl。PCR反应条件:94℃预变性5 min,94℃变性1 min、67℃退火40 s、72℃延伸1 min,共循环35次。PCR产物经1.50%琼脂糖凝胶电泳检测后,行Sanger测序。PCR产物采用p DE1 Directional Expression Kit Ver.2试剂盒(北京擎科生物科技有限公司)进行TA克隆以分析基因变异。
结 果
一、临床表型
先证者(Ⅱ3)体格检查:神志清楚,精神状态正常,发育正常,营养中等;对光反射灵敏,眼动充分,无眼震;双侧鼓腮对称有力,双侧软腭上提受限,悬雍垂居中,伸舌居中,无舌肌萎缩、纤颤,咽反射减弱,吞咽动作缓慢,颈部柔软,四肢肌力和肌张力正常,深浅感觉正常,生理反射正常,病理征呈阴性,脑膜刺激征阴性。实验室检查:血清CK 128 U/L(40~200 U/L),乳酸脱氢酸(LDH)为200 U/L(20~250 U/L),肝肾功能试验和甲状腺功能试验均于正常值范围;新斯的明试验阴性。肌电图和头部MRI检查均未见异常。进一步追问先证者病史及家族史,得知该家系中连续两代成员均有OPMD患者,具有显性遗传模式特点:先证者之父50岁发病,首发症状为眼睑下垂,进行性加重伴吞咽困难,于70岁死亡(死因不详);先证者(Ⅱ3)及其二姐(Ⅱ2)、四弟(Ⅱ4)为子代中发病并生存的患者,发病年龄为53~57岁,平均55.33岁;均以眼睑下垂为首发症状(图2),进行性加重,并于3~5年后出现吞咽困难。该家系中4例OPMD患者的临床资料参见表1。

表1 家系中4例OPMD患者的临床资料Table 1.Clinical data of 4 pedigree patients with OPMD

图2 OPMD家系成员眼肌表现 2a 先证者出现眼睑下垂 2b 先证者之二姐出现眼睑下垂 2c 先证者之大姐眼肌表现正常Figur e 2 Clinical manifestations of ocular muscles in family members of OPMD The proband had drooping eyelids(Panel 2a).The second sister of the proband had drooping eyelids(Panel 2b).The proband's elder sister was normal muscle function(Panel 2c).
二、基因检测结果
该家系中6位家族成员的Sanger测序结果显示,先证者之大姐(Ⅱ1)、六妹(Ⅱ6)PABPN1基因无异常,PABPN1基因外显子1的碱基序列为(GCN)10,即(GCG)(6GCA)(3GCG),共编码6个(GCG)丙氨酸重复序列,TA克隆测序峰图以单峰呈现;其余4位家族成员PABPN1基因外显子1均出现3个(GCG)异常重复扩增,为杂合子(GCN)10(/GCN)1(3图3),其中先证者(Ⅱ3)及其二姐(Ⅱ2)、四弟(Ⅱ4)已出现明显的临床症状,先证者之五妹(Ⅱ5)尚未出现相应临床症状。最终先证者诊断为OPMD,该家系明确为OPMD家系。

图3 PABPN1基因检测结果 3a 正常PABPN1基因型(GCN)10,即(GCG)(6GCA)(3GCG) 3b 先证者存在PABPN1基因杂合突变,可见突变区野生型和突变型等位基因波峰重叠 3c 先证者PABPN1基因经TA克隆测序呈(GCN)13,即(GCG)(9GCA)(3GCG),增加3个(GCG)异常重复扩增(红框所示),其基因型为(GCN)10(/GCN)13Figur e 3 PABPN1 gene detection findings PABPN1 normal genotype(GCN)10:(GCG)6(GCA)3(GCG)(Panel 3a).The PABPN1 gene of the proband was mutated to heterozygous,and the wild-type and mutant allele peaks overlap in the mutant region(Panel 3b).The PABPN1 gene of the proband was sequenced by TA cloning and sequencing showed(GCN)13:(GCG)9(GCA)3(GCG)was abnormally amplified,an increase of 3(GCG),which genotype was(GCN)10/(GCN)13(red box indicates,Panel 3c).
讨 论
本文家系中男女均有发病,发病年龄>50岁,以眼睑下垂为首发症状,进行性加重伴吞咽困难,但肢体肌力和肌张力正常、新斯的明试验阴性,PABPN1基因检测提示基因型为(GCN)10(/GCN)13,结合临床表现和基因检测结果,最终先证者(Ⅱ3)及其二姐(Ⅱ2)、四弟(Ⅱ4)诊断为OPMD,该家系明确为OPMD家系。
OPMD发病隐蔽,多数为显性遗传模式,少数呈隐性遗传模式,发病年龄较晚,眼睑下垂和吞咽困难为主要临床表现,但二者出现时间不同,因此应注意与老年性重症肌无力(MG)、老年性眼睑下垂、眼咽远端型肌病(OPDM)相鉴别:老年性重症肌无力和老年性眼睑下垂的主要症状为眼睑下垂,但老年性重症肌无力新斯的明试验呈阳性;OPDM的致病机制与GIPC1基因5’非翻译区(GCN)异常重复扩增相关[13];但上述3种疾病PABPN1基因检测均正常。
PABPN1基 因 型(GCN)10(/GCN)13广 泛 见 于OPMD家系[4],(GCN)重复扩增数目与临床症状无显著关联性[2,7],但是不同基因型的临床症状存有差异。一项针对法国地区354例OPMD患者的研究发现,其(GCN)重复扩增数目可影响疾病严重程度和进展[14]。有文献报道,眼睑下垂常是携带(GCN)14的男性OPMD患者的首发症状[15]。OPMD患者肌肉受累部位及程度有一定的家族聚集性[16],本文家系均以眼睑下垂为首发症状,进行性加重伴吞咽困难。患者矫正眼睑下垂后可发生严重眼部并发症,推测可能是由于眼球拉伤和眼肌麻痹,暂时性拉绳缝合术是治疗眼部并发症的有效方法[17]。对于吞咽困难的患者,症状改善维持时间是选择手术治疗的关注点[18]。OPMD患者肌肉组织病理学可见两种类型包涵体,一种为核内管状细丝样包涵体,呈栅栏样排列,为OPMD所特有;另一种为管丝样包涵体[19],常用于未行基因诊断前的病理诊断。体外研究和动物实验显示,丙戊酸对OPMD肌细胞和蠕虫模型具有保护作用,提示其治疗有效[20];在OPMD小鼠模型中测试的一种基因疗法,可以恢复PABPN1基因在肌细胞胞核内形成的核内包涵体,并防止肌纤维化和肌萎缩,以使肌肉力量恢复至健康水平,从而展现基因治疗的良好应用前景[21]。
本文家系中具有临床症状的家族成员基因检测显示,PABPN1基因外显子1中3个(GCG)丙氨酸的异常重复扩增的基因型(GCN)10(/GCN)13,从而证实为OPMD家系,提示云南人中存在PABPN1基因变异。既往研究提示,部分地区OPMD的基因变异可能具有“奠基者”效应[22-24],目前我国报道的OPMD患者大多为相同基因型,但临床表现明显不同,进一步表明OPMD具有临床表型异质性[25]。由于目前尚缺乏基因治疗和特异性药物,对症处理或手术治疗是改善临床症状和生活质量的常见方法。通过积累更多地区和人群中的病例临床表型和基因变异数据,并结合肌肉组织活检等临床检测,可以为该病的分子病理学机制、临床诊断与治疗、预后,以及家族遗传咨询提供更全面的信息。
利益冲突无
