不同抗氧化剂条件下生物样品中卡托普利浓度测定方法的建立及应用
2021-09-19赵艳艳王廷春李小川张枢袁小青段小群
赵艳艳 王廷春 李小川 张枢 袁小青 段小群






【摘要】目的在二硫苏糖醇和抗坏血酸两种不同抗氧化剂条件下,建立测定卡托普利血浆浓度的液相色谱-串联质谱法(LC-MS/MS),经方法学验证后将两种方法分别应用于临床样本检测中,系统的对比分析两种抗氧化剂对卡托普利浓度检测的影响,为卡托普利的临床研究提供科学依据。方法:在不同抗氧化剂条件下考察卡托普利的稳定性,添加了抗氧化剂的卡托普利血浆样本,经2,4-二溴苯乙酮衍生化后,乙腈沉淀蛋白处理,ZORBAX SB-C184.6*150mm,3.5μm色谱柱洗脱分离,采用电喷雾离子源多反应监测模式下的正电离法分析检测,建立卡托普利LC-MS/MS方法,并在此基础上分别进行方法学验证,最后将两种方法应用于卡托普利临床生物样本分析中。结果:卡托普利血浆样本在10~2000ng/mL(二硫苏糖醇)和2~1000ng/mL(抗坏血酸)(r均≥0.9994)范围内线性关系良好,批内精密度≤7.4%(n=6),批间精密度≤14.4%(n=18),准确度为94.7~108.3%,提取回收率为86.6~108.4%(n=6),基质效应的RSD为1.1%~3.5%(n=6)。稳定性(冻融循环、长期、短期、冰浴避光)实验中卡托普利的偏差均小于±15%(n=3)。二硫苏糖醇和抗坏血酸条件下卡托普利的AUC0~t分别为(3474.8±741.4)、(380.1±108.9)ng﹒h/mL,AUC0~∞分别为(3725.2±798.9)、(389.0±110.1)ng·h/mL ,Tmax分别为(0.94±0.36)、(0.72±0.17)h,Cmax分别为(877.7±182.8)、(319.8±119.7)ng /mL。结论:0.5%4 mol/L二硫苏糖醇和0.2%4 mol/L抗坏血酸水溶液均可使卡托普利稳定利于检测。两种抗氧化剂条件下卡托普利方法学验证结果均符合生物分析要求,两种检测方法都可应用于卡托普利临床样本分析,但两种方法的卡托普利药代动力学参数有差异性,临床研究可根据实验需求进行选择。
【关键词】抗氧化剂 ;卡托普利;液相色谱-串联质谱法
卡托普利是一种血管紧张素转换酶的特异性竞争性抑制剂,广泛应用于高血压和充血性心力衰竭的治疗,它在血浆中消除半衰期相对较短,口服生物利用度较低(60-75%),与其它高血压药物相比副作用较小。卡托普利结构中有个活性较高的巯基基团,在生物基质中,硫醇类化合物很容易被氧化成二硫化物或与内源性硫醇结合形成二聚体。由于其活性巯基的存在,很难直接测定人体血浆中的卡托普利,为卡托普利的生物分析带来极大的困扰。
在测量卡托普利血浆浓度前,需先添加抗氧化剂保护卡托普利不被氧化,以防止二硫化物的形成。抗坏血酸(Vitamin C, VC)由于其特殊的结构,在各种化学反应中发挥抗氧化剂的作用。通过抗氧化剂自身氧化,使得空气中的氧与抗氧化剂先结合,消耗卡托普利内部与周围环境的氧,从而防止卡托普利被氧化。二硫苏糖醇(Dithiothreitol, DTT) 是一种常用的巯基保护剂,通过硫醇-二硫键交换反应还原分子内或分子间二硫键,可以将转化的二硫二聚体(或共轭物)还原为卡托普利。卡托普利本身的灵敏性低和性能差,不适用于生物医学分析,但适当的衍生化可以通过LC-MS/MS分析方法进行测量。目前并没有任何研究将两种不同的抗氧化剂进行系统的比较并分析这两种抗氧化剂对卡托普利临床样本检测的影响。
本文在VC和DTT两种抗氧化剂条件下分别建立了测定人血浆中卡托普利的LC-MS/MS方法,在此基础上对这两种方法进行了验证,最后成功应用于卡托普利临床样本分析。经过不同方法检测得出的卡托普利浓度体现了较大的差异性,该数据能够有助于判断实际临床试验过程中卡托普利抗氧化剂的选择。
1材料
1.1仪器
液相色谱系统(日本岛津公司,包括DGU-20A3R脱气机,LC-20AD液相泵,SIL-20AC自动进样器,CTO 20S柱温箱);三重四极杆串联API4000质谱仪(配备电喷雾离子源(ESI源)和Analyst 1.6.2数据处理软件(美国AB Sciex公司);医用离心机(3k15,德国Sigma公司);天平(Secura 225D-1CN,德国Sartorius公司);涡旋混合仪(Vortex-Genie2,美国Scientific公司)。1.2药品与试剂
卡托普利(中国食品药品检定研究院,批号:100318-201904,纯度:99.7%)卡托普利-d3(中国食品药品检定研究院,批号:20-JHY-124-3,化学纯度:95%;同位素纯度:98.3%);抗坏血酸(Aladdin,批号:L1920112,纯度:99.99%);二硫蘇糖醇(Aladdin,批号:P1378642,,纯度:98%);2,4-二溴苯乙酮(Aladdin,批号:L1522183,纯度:99%);卡托普利片(美国Bristol-Myers Squibb公司,规格25 mg/片)。
2 方法和结果
2.1不同抗氧化剂前处理方法开发
2.1.1 DTT对卡托普利生物样品稳定性的影响将4 mol/L的DTT水溶液按照5%的比例加入血浆样本中,考察卡托普利血浆样本在冰浴避光0.5 、1 、2 h的稳定性,见表1。结果表明卡托普利血浆样品中加入0.5%4mol/L DTT水溶液后,卡托普利氧化被抑制,RE在-6.8%~-3.0%之间,其血浆样品在0.5、1、2h无明显变化,稳定性较好。
2.1.2DTT对卡托普利稳定性的影响通过不加DTT放置0.5h、1 h后与再加入DTT经前处理后进样对比分析卡托普利血浆样本的稳定性,见表2。结果表明卡托普利血浆样品在室温避光条件下,降解明显,在降解后的样品中加入0.5%4mol/LDTT水溶液后,DTT对降解产物进行了还原,使得稳定性样品与即刻样品的RE在-10.0%~-4.0%之间。
2.1.3VC对卡托普利生物样品稳定性的影响将3 mol/L、4 mol/L的VC水溶液按照2%的比例加入卡托普利血浆样本中,经前处理后考察卡托普利血浆样本在1 h、4 h的稳定性,见表3。结果表明在冰浴避光条件下,含0.2%4 mol/LVC水溶液的卡托普利血浆样品更稳定。
2.2溶液的制备
2.2.1 标准曲线、质控样品工作溶液的制备精密称取卡托普利标准品2份,用甲醇溶液配制成1 mg/ml的卡托普利储备液,1份用于标准曲线工作溶液的的配制,1份用于质控工作溶液的配制。取卡托普利的储备液以50%甲醇水进行稀释,获得卡托普利标准曲线及质控的工作溶液。精密称取对照品卡托普利-d3,用甲醇溶液配制成卡托普利内标储备液并用乙腈溶液配制成内标工作溶液。
2.2.2DTT条件下卡托普利标准曲线、质控血浆样品的制备取标准曲线、质控工作液10μL加入190μL的混合空白血浆(含0.5% 4 mol/L的DTT水溶液),混匀配制成卡托普利质量浓度分别为10.0、25.0、50.0、100、200、500、1000、2000ng/mL标准曲线系列样品和质量浓度分别为10.0 、20.0 、160 、1600ng/mL 的质控样品。
2.2.3 VC条件下卡托普利标准曲线、质控血浆样品的制备取标准曲线、质控工作液10μL加入190μL的混合空白血浆(含2%4 mol/L的VC水溶液),混匀配制成卡托普利质量浓度分别为2.00、5.00、10.0、50.0、100、200、500 1000 ng/mL的標准曲线系列样品和质量浓度分别为2.00、4.00 、300 、800 ng/mL 的质控样品。
2.2.4 血浆样本的处理取200μL血浆样本加入10μL内标工作溶液及40μL 2,4-二溴苯乙酮涡旋混匀1min,室温条件下衍生30min,加入1000μL乙腈,涡旋混匀1min,15400g条件下离心10min,取200μL进样分析。
2.3色谱条件与质谱条件
2.3.1 色谱条件 色谱柱:ZORBAX SB-C18(4.6*150mm,3.5μm,Agilent);预柱:Accucore XL C18(2.1*10mm,4μm,Thermo);流动相:水溶液(含0.1%甲酸和5mmol/L乙酸铵)-乙腈溶液;梯度洗脱((0.01min~0.20min,30-80%B;0.02min~2.00min,80%B;2.00min~2.01min,80-30%B;2.01min~4.00min,30%B);流速:1ml/min;柱温40.0℃;自动进样器温度为4℃;进样体积为2μL。
2.3.2 质谱条件 带有ESI源的API4000质谱,以正离子模式,采用多反应监测(MRM)扫描模式。喷雾电压:5.5kV;喷雾器温度:500℃;雾化气:50psi;加热辅助气:50psi;气帘气:20psi;碰撞气:8psi;扫描时间:200ms。卡托普利经衍生剂2,4-二溴苯乙酮衍生化后含有碱性氮原子,易在ESI源正离子检测模式下产生较高响应,卡托普利衍生化物定量分析质荷比(m/z)414.1→216.0, 卡托普利-d3内标离子对衍生化物定量分析质荷比(m/z)m/z417.1→219.0。
2.4专属性考察
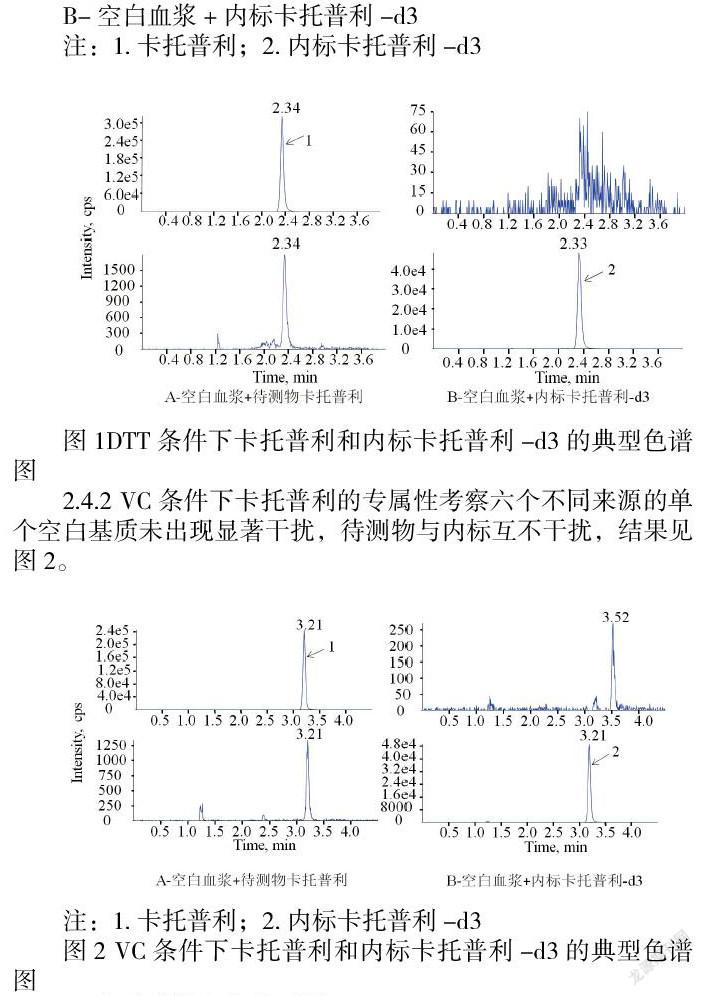
2.4.1 DTT条件下卡托普利的专属性考察六个不同来源的单个空白基质未出现显著干扰,待测物与内标互不干扰,结果见图1。
2.4.2 VC条件下卡托普利的专属性考察六个不同来源的单个空白基质未出现显著干扰,待测物与内标互不干扰,结果见图2。
2.5标准曲线和定量下限
分别取“2.2.2”和“2.2.3”项下标准曲线样品和质控样品,按照“2.2.4”项下方法处理后,进样分析。DTT条件下得到的典型标准曲线为y=0.00455x-0.000773(r=0.9999),卡托普利血药浓度在20~2000 ng/mL范围内线性关系良好,其定量下限为20 ng/mL。VC条件下得到典型标准曲线为y=0.00342x+0.000629(r=0.9999);卡托普利血药浓度在2~1000 ng/mL范围内线性关系良好,其定量下限为2ng/mL。
2.6 准确度与精密度考察不同条件下卡托普利的准确度与精密度考察 取“2.2.2”和 “2.2.3”项下质控样品,按照“2.2.4”项下方法处理后,进样分析,测定3个分析批,每批每个浓度做6个平行样品进样分析。考察其批内及批间的准确度和精密度,结果见表4和表5。
2.7 提取回收率和基质效应考察6批不同来源的空白血浆分别加入VC和DTT两种抗氧化剂经预处理后得到上清液加入质控工作溶液配制成基质效应样品,与相同浓度的纯溶液样品所测得的峰面积比值进行比较,考察其基质效应。提取回收率考察3个质控浓度样品与空白血浆样品预处理后再加入适当工作液配的同浓度样品所测得的峰面积比值。不同条件下卡托普利的基质效应和提取回收率分析物提取回收率在86.6%~108.4%之间,基质效应在93.4%~108.3%之间,CV均≤4.3%;,符合生物样品分析要求,见表6和表7。
2.8稳定性考察 不同条件下卡托普利的稳定性考察按“2.6.1”项下方法配制低、高质量浓度的卡托普利质控样品,每个浓度平行3份,考察血浆样品中卡托普利在冰浴避光条件下短期、-25 ℃避光放置7天、-80 ℃避光放置28天、60天、5次反复冻融、处理后样品自动进样器4 ℃放置48 h的稳定性,以上条件下稳定性均良好,结果见表8和表9。
2.9药动学实验
受试者给药前一天晚上禁食不禁水10小时以上,给药当天空腹口服一片卡托普利片(25 mg),给药前后lh内禁止饮水。给药前和给药后15min、30min、45min、l.00 h、l.25h、l.50h、l.75h、 2.00h、 2.50h、3.00h、4.00h、5.00h、6.00h、8.00h、10.00h、12.00h、24.00h共18个采血点,每个采血点采4 mL,4000 r/min离心5 min,分离血浆分别置于提前加好不同抗氧化剂的冻存管中,-80 ℃保存,后取血浆样品按“2.2.4”项下方法处理后,进样分析,采用Phnenix WinNonlin 7.0软件以非房室模型计算临床受试者体内卡托普利的Cmax,AUC0-t,AUC0-∞,Tmax,t1/2,λz,AUC_%Extrap等主要药动学参数,药动学参数结果见表10,平均药时曲线图见图3和图4。
3讨论
为了考察两种不同抗氧化剂对卡托普利生物样品检测的影响,本研究建立了DTT和VC两种不同条件下生物样品中卡托普利的LC-MS/MS分析方法,并对这两种方法进行了验证,均符合生物分析要求,这两种分析方法均可用于卡托普利临床样本检测,通过不同方法对临床样本进行检测,所得结果体现了差异性。
将DTT和VC两种抗氧化剂条件下的LC-MS/MS分析方法应用到临床样本检测时,我们发现DTT条件下卡托普利血药浓度明显比VC条件下的卡托普利血药浓度高,因为DTT将卡托普利二硫化物还原为卡托普利,实际检测的浓度为卡托普利和卡托普利二硫化物的总浓度,因此DTT条件下卡托普利血药浓度偏高。DTT条件下卡托普利达峰时间较晚,半衰期较长。
本研究建立的两卡托普利检测种方法简单、易于操作,灵敏度高,得出了不同抗氧化剂条件下卡托普利样本浓度及药代动力学的差异性。此项研究为卡托普利临床研究抗氧化剂的选择和卡托普利检测提供了数据支持和参考依据。
参考文献
[1] I Jiménez-Martínez, AM Domínguez-Ramírez, Villafuerte-Robles L . Effect of antioxidants on captopril floating matrices[J]. Pharmaceutical Development and Technology, 2010, 15(3):230-240.
[2] Gan Z , Huang D , Jiang J , et al. Captopril alleviates hypertension-induced renal damage, inflammation, and NF-kB activation[J]. Brazilian Journal of Medical & Biological Research, 2018, 51(11).
[3] Dortunc, Betul, Ozdemir, et al. Pharmacodynamical evaluation of matrix transdermal type transdermal therapeutic systems containing captopri [J]. Acta Poloniae Pharmaceutica: Durg Research, 2015, 72(4):799-806.
[4] Mahmoud W , Kuemmerer K . Captopril and its dimer captopril disulfide: Photodegradation, aerobic biodegradation and identification of transformation products by HPLC–UV and LC–ion trap-MSn[J]. Chemosphere, 2012, 88(10):1170-1177.
[5] 刘亚君. 卡托普利间接测定方法的建立及黄秋葵中GSH和总巯基(-SH)的检测[D]. 延边大学, 2016.
[6] Iqbal F M , Ahmad M , Zubair M M , et al. Determination of Captopril in Plasma by High-Performance Liquid Chromatography: Application in an In-Vivo Evaluation of Drug Release from Hydrogel[J]. LATIN AMERICAN JOURNAL OF PHARMACY, 2015, 34(5).
[7] Salem I I , Saif W A , Jmeian Y , et al. A selective and rapid method for the quantification of captopril in human plasma using liquid chromatography/selected reaction monitoring mass spectrometry[J]. Journal of Pharmaceutical and Biomedical Analysis, 2005, 37(5):1073-1080..
[8] Bojarska J , Maniukiewicz W , A Fruziński, et al. Captopril and its dimer captopril disulfide: comparative structural and conformational studies[J]. Acta Crystallographica Section C Crystal Structure Communications, 2015, C71(3):199-203.
[9] 陳涛,钟勘,薛彩福,杨蓓,陈笑艳,钟大放,朱晓彤,杨勇.柱前衍生化LC-MS/MS测定生物等效性研究受试者血浆中卡托普利的浓度[J]. 中国新药杂志,2020,29(02):176-182.
[10] Vancea S , Imre S , G Donáth-Nagy, et al. Determination of free captopril in human plasma by liquid chromatography with mass spectrometry detection[J]. Talanta, 2009, 79(2):436-441.
[11] Olabisi A , Wimalasena K . Rational approach to selective and direct 2-O-alkylation of 5,6-O-isopropylidine-L-ascorbic acid[J]. Journal of Organic Chemistry, 2004, 69(21):7026.
[12] Macan A M , Kraljevi T G , Rai-Mali S . Therapeutic Perspective of Vitamin C and Its Derivatives[J]. Antioxidants, 2019, 8(8):247.
[13] 葛颖华,钟晓明.维生素C和维生素E抗氧化机制及其应用的研究进展[J].吉林医学,2007(05):707-708.
[14] Tian Y , Cao J , Luo L , et al. Determination of Zofenopril and Its Active Metabolite in Human Plasma Using High-Performance Liquid Chromatography Combined With a Triple-Quadruple Tandem Mass Spectrometer[J]. Journal of Chromatographic Science, 2015(2):253-62.
[15] 王玉,王靜,刘力力,刘冬,陈镕,李瑞飞,魏春敏,杨进波. 卡托普利片生物等效性研究基本考虑[J]. 中国临床药理学杂志,2020,36(12):1603-1605.
[16] Musharraf S G , Bhatti M S , Choudhary M I , et al. Screening of inhibitors of angiotensin-converting enzyme (ACE) employing high performance liquid chromatography-electrospray ionization triple quadrupole mass spectrometry (HPLC-ESI-QqQ-MS)[J]. European Journal of Pharmaceutical Sciences, 2017, 101:182-188.
[17] 施建丰, 姚孝明, 王志国,等. 优化的液相色谱-串联质谱联用法测定人血浆中卡托普利的浓度及其药动学研究[J]. 中国医院用药评价与分析, 2013, 013(006):534-536.
