HPLC法测定芪蓝囊病饮中的靛玉红
2021-09-16牛华星章安源李有志尹伶灵刘少宁魏秀丽张志民张传津
牛华星,章安源,赵 见, 郭 腾,李有志,尹伶灵,刘少宁,陈 玲,魏秀丽,张志民,张传津*
(1.山东省兽药质量检验所,济南 250022;2.日照市东港区涛雒计生委,日照 276800)
芪蓝囊病饮是收载于《兽药质量标准》2017卷的纯中药制剂[1],主要成分为黄芪、板蓝根、大青叶、地黄、赤芍,具有提高疫苗免疫应答效果(参考说明书的功能主治)。目前本制剂标准只是对黄芪、板蓝根、大青叶进行定性薄层鉴别,缺少对有效成份定量控制。有文献表明[2-3],靛玉红(indirubin)是大青叶、板蓝根的主要指标成分,因此本文主要开展了反相高效液相色谱法测定靛玉红方法学研究,为完善芪蓝囊病饮的质量标准提供依据。
1 实验部分
1.1 仪器与试药 Waters e-2695(配二极管阵列检测器2998);靛玉红对照品(批号:110717-200204)(中国药品生物检定所);乙腈(色谱纯);磷酸(分析纯,国药集团化学试剂有限公司);水为超纯水;供试品:芪蓝囊病饮口服液(某公司提供,批号:202006271,202006281,202006291,202007271,202007281,202007291),阴性空白制剂自制;其他试剂均为分析纯。
1.2 方法与结果
1.2.1 色谱条件 色谱柱为Waters Symmetry C18柱(250 mm×4.6 mm×5 μm);流动相A:乙腈;流动相B:0.2%磷酸水溶液;流速:1.0 mL/min;进样量:10 μL;柱温:35 ℃。检测波长为289 nm,梯度洗脱条件见表1;

表1 梯度洗脱条件
1.2.2 溶液的制备 取13.58 mg靛玉红对照品,精密称定,加三氯甲烷配制成1358 μg/mL的对照品储备液。取1 mL储备液用甲醇制成67.9 μg/mL标准工作液,依次倍比稀释成系列浓度:2.0、1.5、1.0、0.8、0.6、0.5 μg/mL标准工作液。
取1 mL供试品溶液于50 mL,离心管中,加入9 ml三氯甲烷,振摇1 min,静置分层弃去上清液,0.22 μm微孔滤膜滤过,即得。另取不含大青叶的阴性空白制剂,同法制成阴性空白对照溶液。
1.2.3 专属性试验 分别吸取10 μL“1.2.2”项下阴性空白对照溶液、供试品溶液、对照品溶液,按“1.2.1”项下色谱条件,注入液相色谱仪,记录色谱图。在靛玉红对照品相应色谱峰位置处无其他色谱峰出现(图1)。
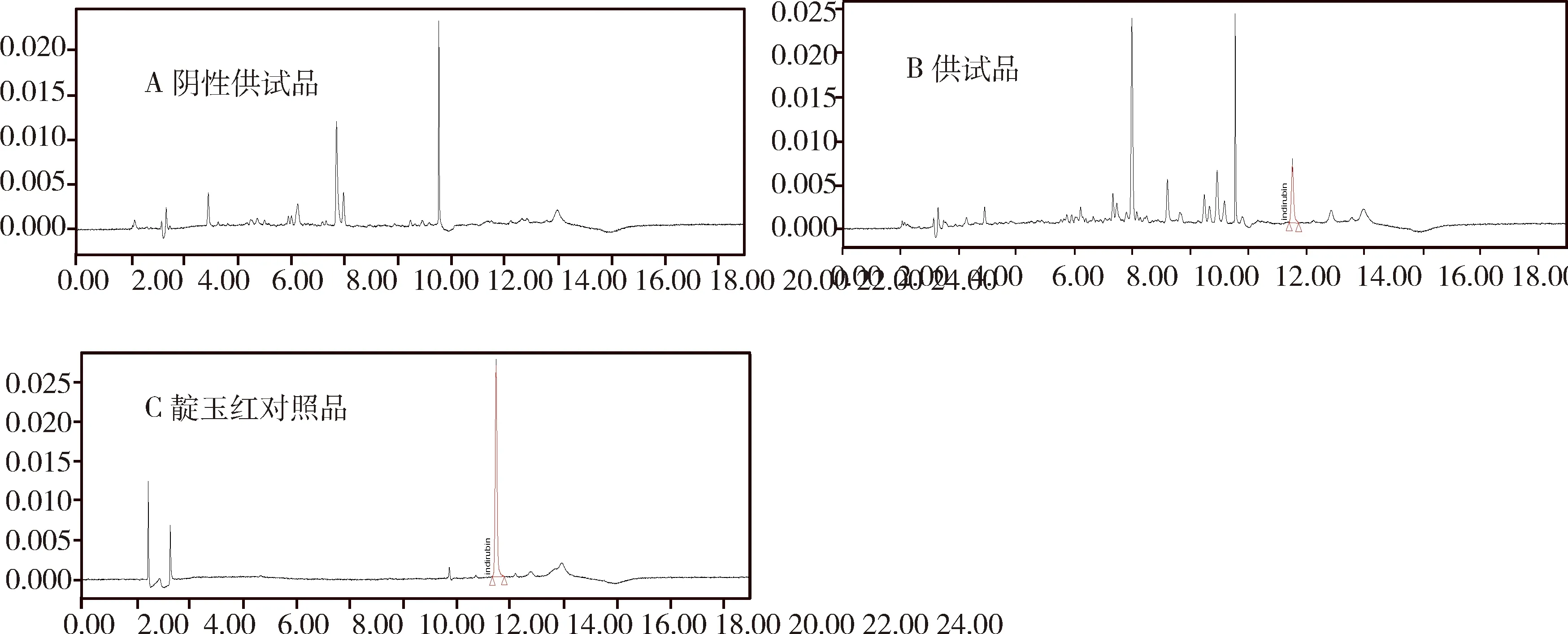
图1 阴性供试品溶液(A)供试品溶液(B)靛玉红对照品溶液(C)
1.2.4线性关系考察 分别吸取“1.2.2”项下标准曲线工作液,在“1.2.1”项色谱条件下,使用梯度洗脱方法,可以有效分离目标峰,靛玉红在0.5~2.0 μg/mL范围内呈良好的线性关系(图2)。
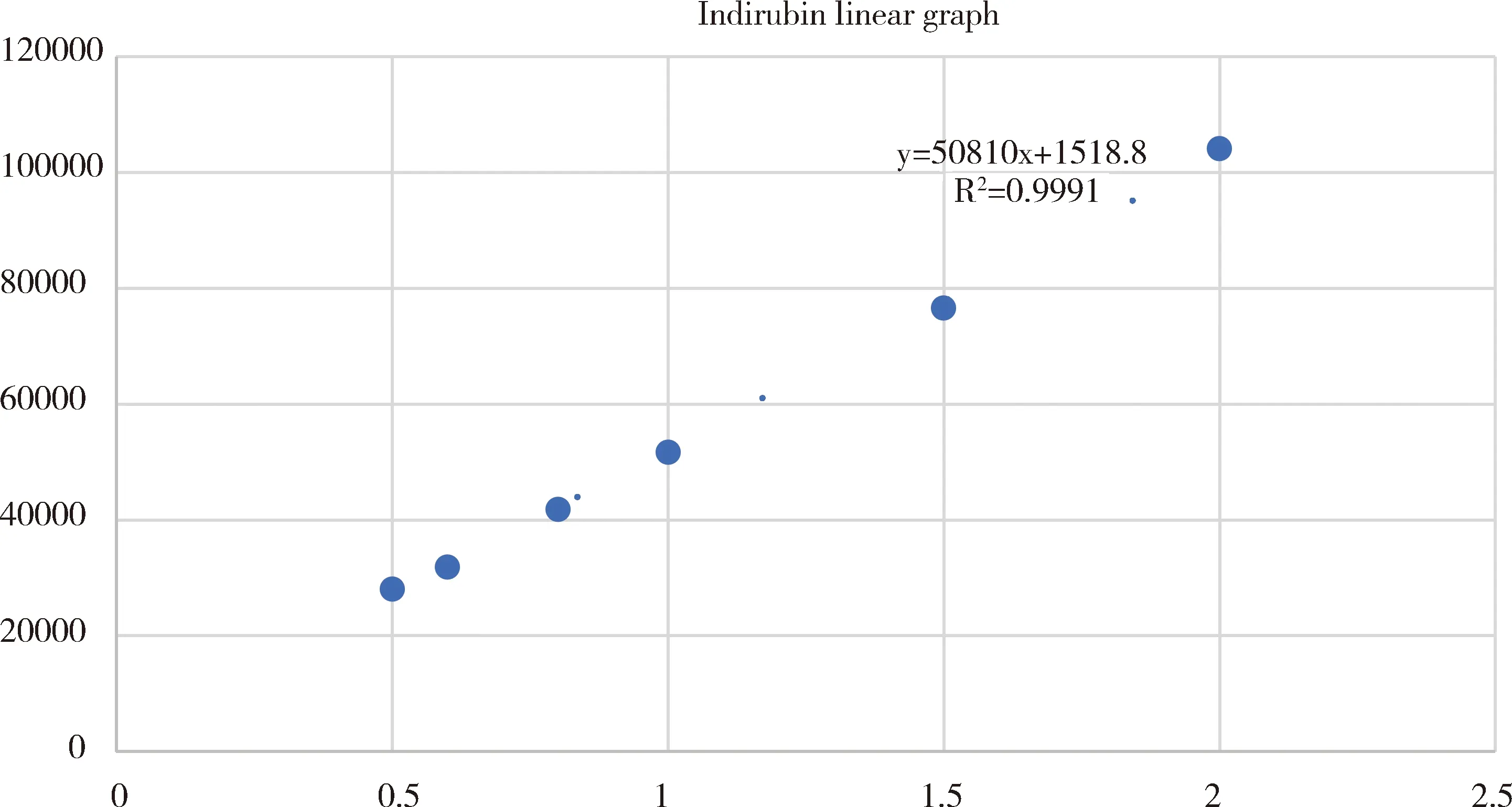
图2 靛玉红溶液的标准曲线
1.2.5 重复性与稳定性 精密量取同批供试品1 mL,按“1.2.2”项下方法制备供试品溶液6份,注入液相色谱仪,进样10 μL,记录色谱图,靛玉红峰面积RSD为 1.4 %。说明样品重复性良好。
将其中一份供试品溶液,取10 μL分别于放置后2、5、7、12 h注入液相色谱仪,记录色谱图,靛玉红峰面积RSD为1.4%。结果表明供试品溶液在12 h内稳定性良好。
1.2.6 精密度试验 取“1.2.2”项下靛玉红对照品溶液,注入液相色谱仪,进样10 μL,连续进样6次,记录色谱图,靛玉红峰面积RSD为0.4 %。结果表明,仪器精密度良好。
1.2.7 加样回收实验 精密量取已知含量的样品1 mL,精密加入10.1850 μg/mL的靛玉红对照品0.5 mL,制备阳性添加供试品6份。按“1.2.2”项下方法供试品溶液,进样10 μL,注入液相色谱仪,记录色谱图,计算加样回收率计算加样回收率(表2)。回收率在89.2%~92.4%之间,RSD为1.34%。表明该方法回收率好,准确度满足检测要求。
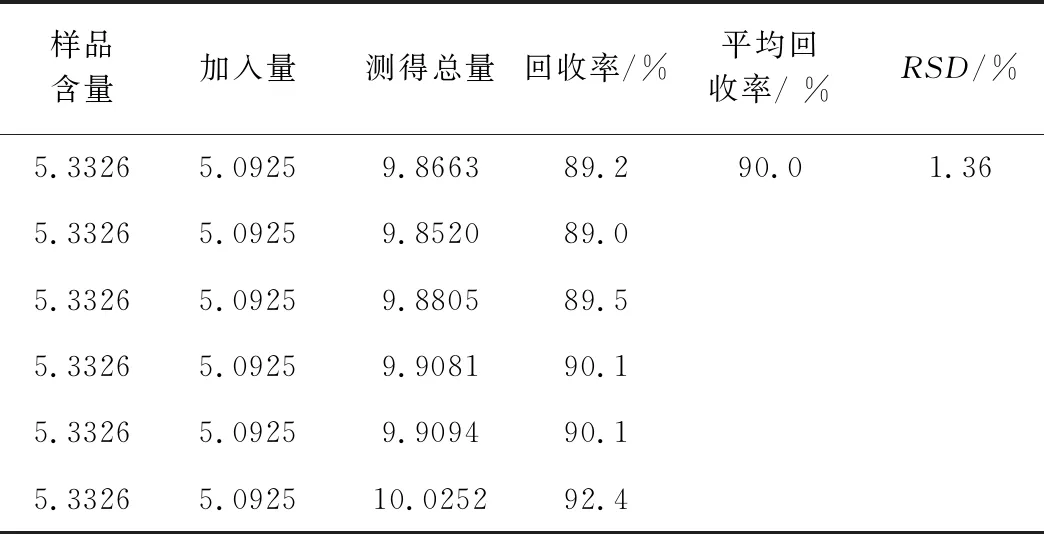
表2 加样回收率试验结果(μg/mL,n=6)
1.2.8 样品的测定 精密量取6批芪蓝囊病饮进行测定。分别吸取供试品溶液和对照品溶液各10 μL,按“1.2.1”项下色谱条件,外标法计算含量。结果测定6批芪蓝囊病饮中靛玉红的含量(表3)。
1.2.9 不同仪器相同色谱条件下样品检测比较 使用waters e2695和agilent高效液相色谱仪,按“1.2.1”项下色谱条件运行,对出峰时间和含量进行比较(表3),结果表明:芪蓝囊病饮中靛玉红的平均含量,相对偏差1.52%。结果表明,该方法在不同仪器上,检测结果稳定。
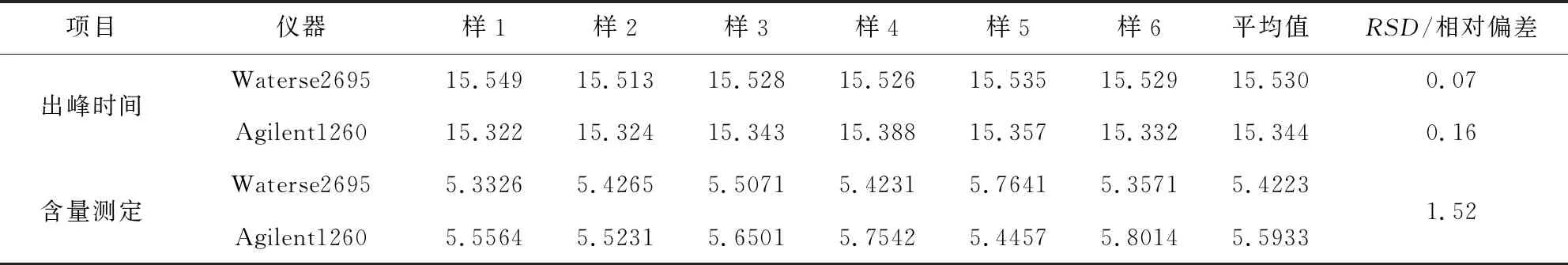
表3 不同仪器相同色谱条件结果表(μg/mL,n=6)
2 讨论与结论
2.1 指标成分的选取[4-6]板蓝根、大青叶是芪蓝囊病饮的主要药成分。板蓝根别名靛青根、蓝靛根、靛根,是十字花科菘蓝属植物菘蓝的干燥根,含有靛玉红、吲哚苷、多种氨基酸、β-古甾醇、植物性蛋白、糖类等成分。大青叶是十字花科菘蓝属植物菘蓝的干燥叶,含有靛玉红、靛蓝、菘蓝苷B、扶桑甾醇、芥苷类多种成分。其中靛玉红为板蓝根、大青叶中含量较高的主要成分,化学研究表明靛玉红可作为药材和其制剂的质量控制指标。大青叶中另一含量较高的成分为靛蓝,靛蓝与靛玉红为同分异构体,靛蓝分子中的氢键小于靛玉红,其性质不稳定,不耐光和热。本文只选择靛玉红作为指标成分进行实验。
2.2 提取溶剂的选择 根据文献相关方法[7-9],实验比较了甲醇,乙醚,氯仿,二甲基甲酰胺等溶剂的提取效果。采用“1.2.1”色谱条件进样,比较峰面积值,结果显示,甲醇提取效果较差,二甲基甲酰胺提取效果较好,靛玉红在乙醚,氯仿中提取效果最好,因乙醚对c18色谱柱有一定损害,最终选择氯仿为供试品提取溶剂。
2.3 提取方式选择 实验比较了不同提取方式对制剂中靛玉红提取效率的影响。精密量取芪蓝囊病饮供试品1 mL于玻璃试管中,加入9 mL氯仿,分别采取,振摇,超声。采用“1.2.1”色谱条件进样,比较峰面积值,结果显示,振摇,超声的提取效率基本一致。为方便实验操作,选择振摇作为本方法测试药物的提取方式。
2.4 流动相的选择 根据文献相关方法[10-11],大青叶,青黛和小儿清咽颗粒中靛玉红的采用不同比例的甲醇水或乙腈水为流动相分别测定,在此基础上探讨对比不同的流动相及流动相比例,对芪蓝囊病饮中的靛玉红进行测定。选用甲醇-水(75∶25,V∶V),(80∶20,V∶V);甲醇-1%醋酸(50∶50,V∶V),乙腈-水(53∶47,V∶V)四种流动相,比较色谱图,结果发现乙腈-水(53∶47,V∶V),靛玉红的峰形较好,但是仍与供试品溶液其他成分峰不易分离。本实验最终将流动相优化为乙腈-0.2%磷酸水,并进行梯度洗脱,发现供试品中靛玉红与其他成分分离较好。在waters e2695(PDA)与Agilent1260 infinityⅡ(PDA)上,多次进样,专属性好,满足实验要求。
2.5 影响含量测定的因素 影响芪蓝囊病饮中靛玉红的含量差异因素很多,原料药材产地、采收期、投料和加工工艺,都会使靛玉红的含量有所差异。所以,对制剂中有效成分进行定量控制非常必要。
研究建立了HPLC法测定芪蓝囊病饮中靛玉红的含量,经方法学验证,该方法前处理简单,色谱分离较好,可为完善芪蓝囊病饮的质量标准提供依据。
