中药制剂归芎益母散的质量标准研究
2021-09-16吴雪琴王胜义吴亮红崔东安
吴雪琴, 孙 研,王胜义,王 磊,吴亮红,岳 鹏,崔东安
(中国农业科学院兰州畜牧与兽药研究所甘肃省中兽药工程技术研究中心,中国农业科学院兰州兽医研究所,甘肃 兰州 730050)
归芎益母散是以中兽医学理、法、方、药为基础,遵循君臣佐使原则,结合中药研究,研制出的复方散剂,是由益母草、当归、川芎、红花、香附和甘草[3]等中药制成的散剂,益母草对子宫有双向调节作用,可以降低产后子宫的TNF-α、基质金属蛋白酶抑制物 (TIMP-1) 水平, 启动止血修复机制, 并加快细胞外基质 (ECM) 降解, 从而加速产后子宫修复[1-2]。当归、川芎和红花是中兽医经典活血化瘀药材,对子宫均有调节作用,可加速子宫内残留蜕膜及分泌物排出,促进产后子宫修复[4-6]。甘草具有抗炎、抗氧化、抗肿瘤、抗菌、抗病毒等多种作用[7]。其临床试验验证,功效主要有“活血化淤、行气止痛”,主要用于奶牛产后恶露不行,胎衣不下,血瘀腹痛等症[6]。
为探索归芎益母散的质量控制指标及方法,采用薄层色谱法对归芎益母散中的中药成分进行定性鉴别,采用高效液相色谱法对 “归芎益母散”中的盐酸水苏碱进行定量测定等多方面研究归芎益母散的质量标准。从而保证该中兽药复方制剂的产品质量,使其在临床安全有效使用,以此达到减少奶牛因胎衣不下而引发的各种相关疾病,提高奶制品质量,促进我国奶牛养殖业的发展的目标。
1 材料与方法
1.1 材料
1.1.1 仪器 蒸发光检测器高效液相色谱仪(安捷伦科技有限公司);点样仪(SP-20E,上海科哲有限公司);薄层色谱成像仪(Goodlook-1000,上海科哲有限公司);微量分析天平(RADWAG Wagi Elektroniczne);电子天平(AL104,梅特勒有限公司);超声波清洗器(昆山洁力美超声仪器有限公司);硅胶G板(200 mm×100 mm,国药集团化学试剂有限公司);电热套(上海瑞兹仪器设备有限公司)。
1.1.2 药品 归芎益母散(中国农业科学院兰州畜牧与兽药研究所研制)。盐酸水苏碱(批号:110712-201614,中国食品药品检定研究院)、益母草(120912-201209,中国食品药品检定研究院)当归(120927-201315,中国食品药品检定研究院)、川芎(120918-201411,中国食品药品检定研究院)、红花(120907-201412,中国食品药品检定研究院)。
甲醇、乙腈为色谱纯;丙酮、正己烷、乙醇、盐酸、硫氰酸铬铵、丙酮、硫酸银、氯化钡、冰醋酸等为分析纯。
1.2 方法
1.2.1 定性鉴别
1.2.1.1 益母草的TCL 样品溶液的制备:取归芎益母散3.5 g置锥形瓶中,再加入50 mL无水乙醇,摇匀,超声处理30 min,滤过,滤液蒸干,残渣加0.1 moL/mL盐酸溶液10 mL使溶解,滤过,滤液加入新配制的雷氏铵盐饱和溶液15 mL,4 ℃放冷1 h,滤过,沉淀加约20 mL水洗涤,滤过,加丙酮2 mL使溶解,作为供试品溶液。取其他批次的样品做重复。
阴性对照溶液的制备:根据处方比例,去除益母草后,将各个药按比例称取,按照归芎益母散样品溶液处理方法,制成阴性对照溶液。
对照药材溶液的制备:称取对照药材益母草1.0005 g置锥形瓶中,加10 mL无水乙醇,摇匀,同法制成对照药材溶液。
对照品的制备:精密称取对照品盐酸水苏碱10.0006 mg置5 mL容量瓶中,加甲醇定容,即为2.0003 mg/mL的对照品溶液。
测定方法:参照薄层色谱法(附录43页)试验[8],分别取10 μL阴性对照溶液、三批不同批次的样品溶液,5 μL对照药材溶液、对照品溶液,依次点于硅胶G板上,将丙酮:无水乙醇:盐酸(10∶6∶1)混合液作为展开剂,展开,取出。于105 ℃加热15 min,放冷后喷以稀碘化铋钾试液:三氯化铁试液(10∶1),晾干,日光下检视。
1.2.1.2 当归、川芎的薄层鉴别 样品溶液的制备:取归芎益母散5 g置锥形瓶中,加正己烷50 mL,超声处理30 min,过滤后,将滤液蒸干后,加1 ml正己烷溶解残渣,即为样品溶液。取其他批次的样品做重复[8]。
阴性对照溶液的制备:根据处方比例,去除当归和川芎后,将各个药按比例称取,按照归芎益母散样品溶液处理方法,制成阴性对照溶液。
对照药材溶液的制备:取当归对照药材1 g,川芎对照药材1 g,当归、川芎混合对照药材1 g(当归对照药材、川芎对照药材各0.5 g),按照归芎益母散样品溶液处理方法,制成对照药材当归、川芎以及当归和川芎混合溶液。
方法测定:参照薄层色谱法(附录43页)试验[8],分别取3 μL阴性对照溶液、三批不同批次的样品溶液,2 μl当归和川芎混合对照药材溶液、当归对照药材溶液、川芎对照药材溶液,依次点于硅胶G板上,将石油醚(60~90 ℃):乙酸乙酯(10∶0.4)作为展开剂,展开,取出后晾干,并置紫外灯(365 nm)检视。
1.2.1.3 红花的薄层鉴别 样品溶液的制备:取归芎益母散10.0000 g置锥形瓶中, 再加80%丙酮50 mL,摇匀后超声处理15 min,过滤,蒸干滤液,加5 mL 80%丙酮溶解残渣,作为样品溶液[9-10]。取其他批次的样品做重复。
阴性对照溶液的制备:根据处方比例,去除红花后,将各个药按比例称取,按照归芎益母散样品溶液处理方法,制成阴性对照溶液。
对照药材溶液的制备:称取对照药材红花0.5003 g置锥形瓶中,加入5 mL 80%丙酮,摇匀后超声处理15 min,静置,取上清,作为对照药材溶液。
方法测定:参照薄层色谱法(附录43页)试验[8],分别取10 uL阴性对照溶液、三批不同批次的样品溶液, 5 μL对照药材溶液,依次点于硅胶G薄层板上,将乙酸:乙酯丁酮:甲酸:水(5∶3∶1∶1)混合液作为展开剂,展开后,取出,晾干,日光下检视。
1.2.2 归芎益母散中盐酸水苏碱含量测定
1.2.2.1 溶液的制备 样品溶液的制备:精密称取归芎益母散2.0002 g置圆底烧瓶中,加50 mL 70%乙醇后称重,回流2 h,放冷,70%乙醇加足重量,离心,精密吸取上清液25 mL, 蒸干,用0.1 moL/mL稀盐酸25 mL溶解,离心(3000转/分) 10 min,倾出上清液,加少量0.1 moL/mL稀盐酸洗涤沉淀,洗液合并入上清液,并加入新配制的2.5%硫氰酸铬铵溶液20 mL,振摇,置冰浴(4 ℃)中3 h,离心(3000转/分) 10 min,弃去上清液,沉淀用少量冰水洗涤后,弃去洗液,加2 mL丙酮使溶解。丙酮液中滴加0.5%硫酸银溶液6 mL,离心(3000转/分) 10 min,倾出上清液,沉淀用少量丙酮洗涤,合并洗液与上清液并滴加1%氯化钡溶液2 mL,离心(3000 转/分)10 min,倾出上清液,沉淀用70%乙醇洗涤,洗液加入上清液,蒸干,加适量70%乙醇使其残渣溶解,并转移至5 mL量瓶中,定容,即得归芎益母散样品溶液。
对照品溶液的制备:精密称取3.375 mg对照品盐酸水苏碱置10 mL容量瓶,加甲醇溶解并定容,即得0.0375 mg/mL的盐酸水苏碱对照品溶液。
阴性对照液的制备:根据处方比例,去除益母草后,将各个药按比例称取,按照归芎益母散样品溶液处理方法,制成阴性对照溶液。
1.2.2.2 色谱条件 色谱柱:Venusil HILIC(250 mm×4.6 mm,5 um,天津博纳艾杰尔科技有限公司);乙腈:0.2%乙酸水溶液(83:17);体积流量0.8 mL/min;蒸发光散射器;柱温25 ℃;检测器温度50 ℃,雾化室温度80 ℃;进样量10 uL。理论塔板数按盐酸水苏碱峰计算均不低于5000。
1.2.2.3 专属性试验 分别吸取1.2.2.1中对照品盐酸水苏碱溶液、归芎益母散样品溶液、去除益母草的其他药阴性对照液各10 μL进样,记录色谱峰。
1.2.2.4 线性范围考察 分别精密称定盐酸水苏碱对照品8.115 mg置10 mL容量瓶,加甲醇溶解并定容,摇匀,制成含盐酸水苏碱为0.8115 mg/mL的对照品储备溶液。分别精密吸取对照品储备液适量,加甲醇稀释成含盐酸水苏碱0.115、0.40575、0.202875、0.1014375、0.05071875 mg/mL的系列溶液,精密吸取10 μL,按照上述色谱条件注入色谱仪,以测定峰面积对数对盐酸水苏碱浓度进行回归分析。
1.2.2.5 精密度试验 取盐酸水苏碱对照品浓度为0.285 mg/mL溶液,按2.1.2.2色谱条件连续进样6次,记录峰面积,并计算RSD。
1.2.2.6 日间稳定性考察 取2.1.2.1中样品溶液,按2.1.2.2色谱条件分别在0、4、8、12、24 h吸取该样品溶液10 μL,进样、测定并记录峰面积,并计算RSD。
1.2.2.7 重复性试验 精密称定取同一批样品6份2.0 g,按“样品溶液制备”项下操作,按2.1.2.2色谱条件进样测定,记录峰面积,并计算RSD。
1.2.2.8 准确性试验 取已知含有量的样品6份,精密称定2 g,按样品已知含量的100%加入盐酸水苏碱标准品1.116 mg/mL各加3.3 mL。按2.1.2.1 “样品溶液制备”项下,按2.1.2.2色谱条件进样测定,记录峰面积后计算加样回收率,并计算RSD。
2 结果与分析
2.1 定性鉴别结果
2.1.1 益母草的薄层鉴别 益母草的定性鉴别结果见图1,图中,归芎益母散样品在与盐酸水苏碱对照品相应的位置上,显示相同颜色的斑点,阴性对照样品无相应斑点,但阴性对照样品与益母草对照药材有相同颜色的斑点。参考女金丸、产康复颗粒、四物益母丸等制剂的质量标准中益母草的鉴别[9],仅保留盐酸水苏碱对照品作为鉴别对照,方法具有专一性。

图1 归芎益母散中益母草的的TCL图
2.1.2 当归、川芎的薄层鉴别 当归、川芎的薄层鉴别结果见图2。图中,归芎益母散样品在与对照药材相应的位置上,显示相同的颜色的荧光斑点。

图2 归芎益母散中当归、川芎的TCL
2.1.3 红花的薄层鉴别 红花的薄层鉴别结果见图3。图中,归芎益母散样品在与对照药材相应的位置上,显示相同的颜色的主斑点。阴性样品无相应的斑点,斑点清晰,方法具有专一性。
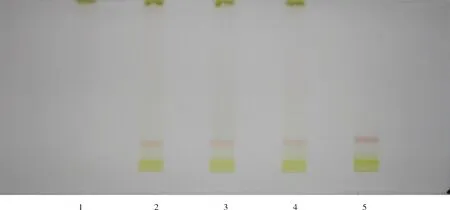
图3 归芎益母散中红花的TCL图
2.2 归芎益母散中盐酸水苏碱含量测定结果
2.2.1 专属性试验结果 试验结果中,样品中盐酸水苏碱峰型对称度好,阴性样品在盐酸水苏碱处无吸收,无干扰归芎益母散的测定。理论塔板数按盐酸水苏碱计为9698。试验结果见图4-图6。
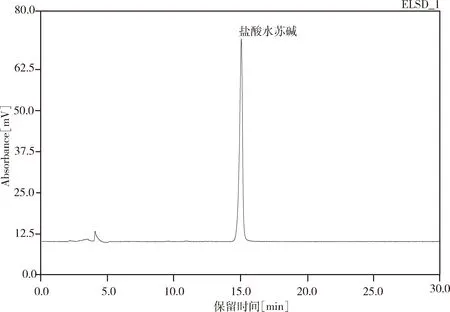
图4 盐酸水苏碱对照品色谱图
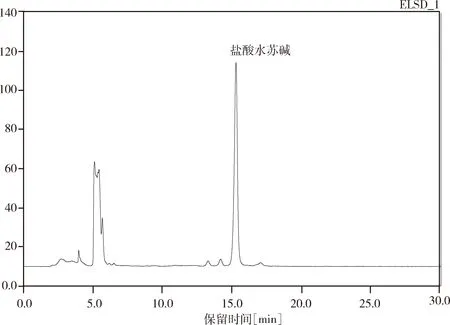
图5 归芎益母散供试品中中盐酸水苏碱色谱图
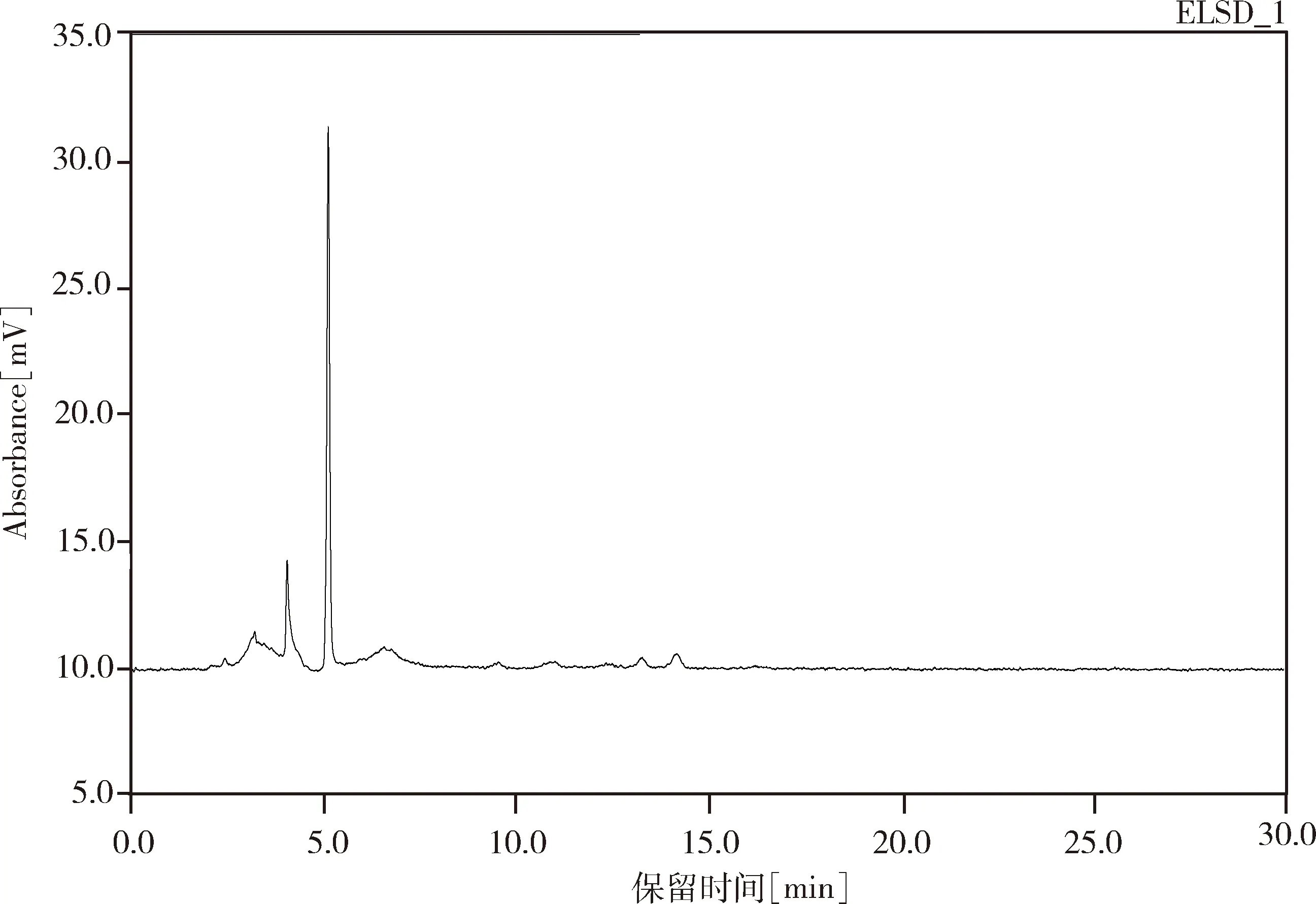
图6 阴性样品中盐酸水苏碱色谱图
2.2.2 线性范围考察 以盐酸水苏碱对照品浓度对数为横坐标(x),色谱峰面积对数为纵坐标(y),进行线性回归,得出回归方程y=1.54x+0.5123,R2=0.9996,见图7,结果表明,盐酸水苏碱浓度在0.0507~0.8115 mg范围与峰面积对数呈良好的线性关系。
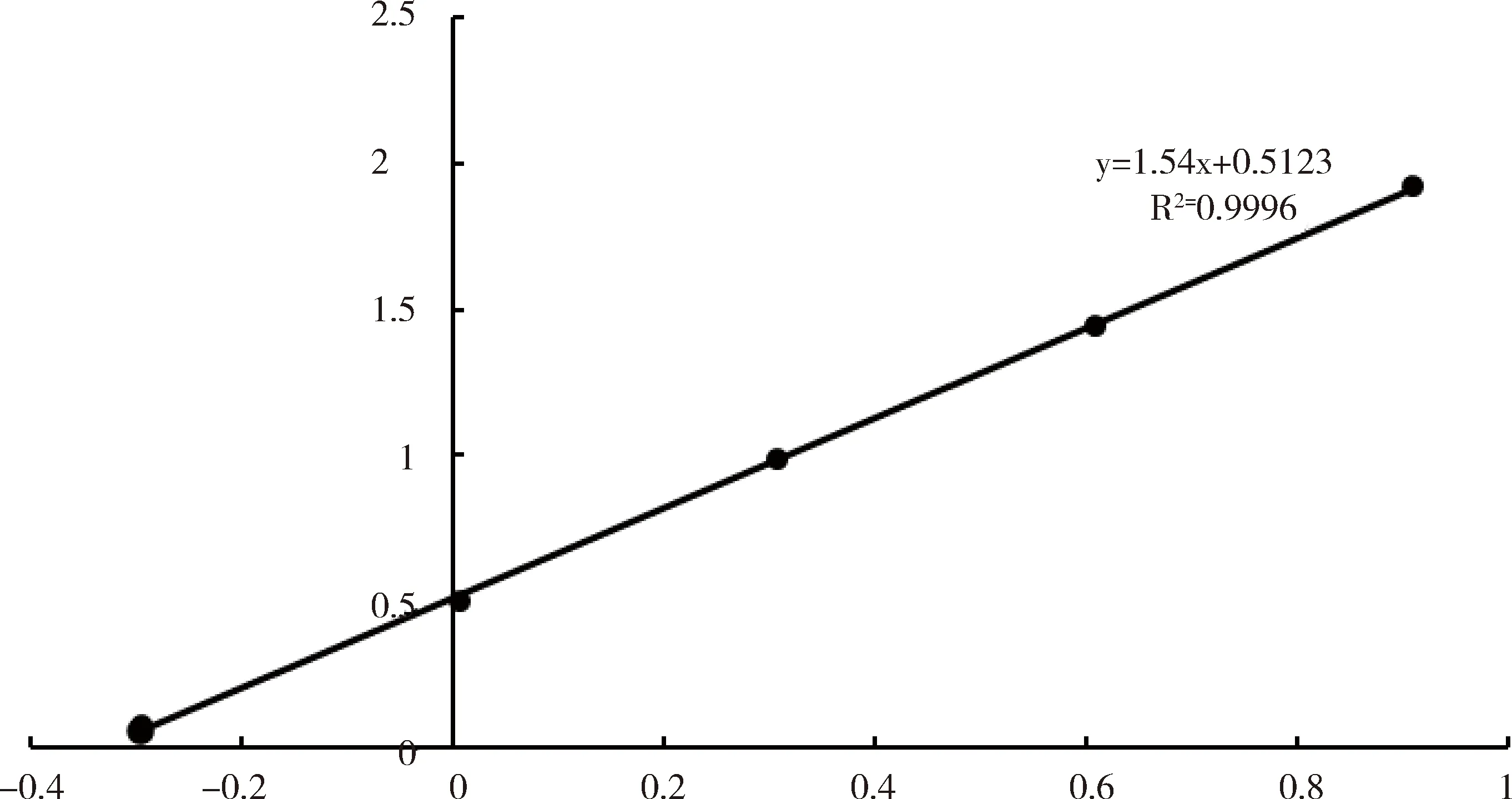
图7 标准曲线
2.2.3 精密度试验 计算RSD,RSD(n=6)为0.83%,表明本方法精密度良好,可以满足含量测定要求。试验结果见表1。
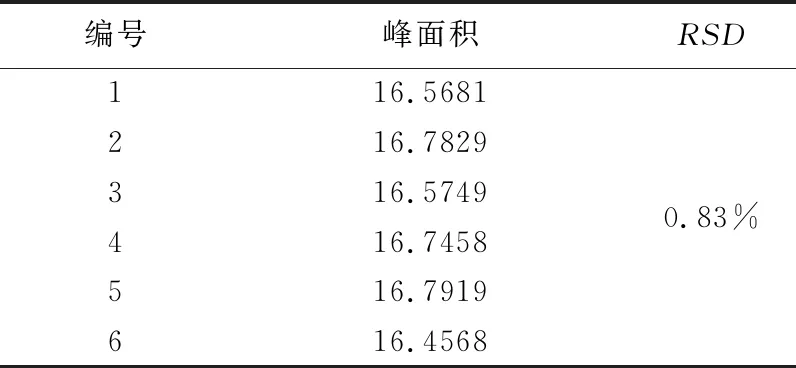
表1 盐酸水苏碱精密度试验结果表
2.2.4 日间稳定性试验 计算含量和RSD,RSD(n=6)为0.66%,表明样品在24 h内稳定,试验结果见表2。
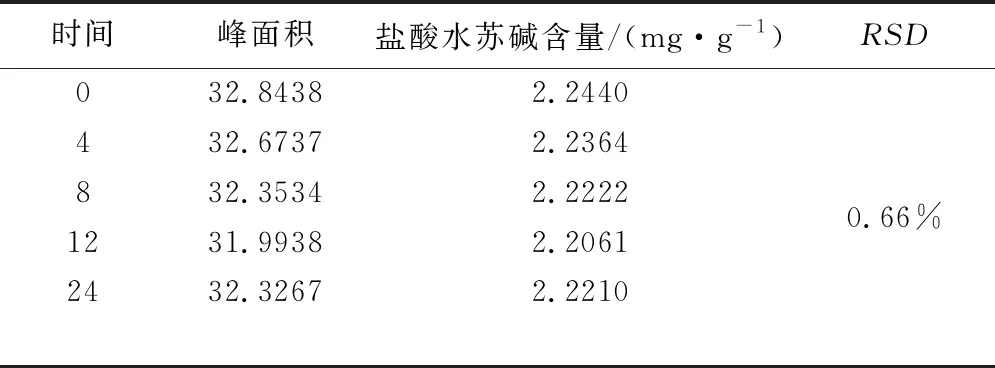
表2 盐酸水苏碱日间稳定性试验结果表
2.2.5 重复性试验 计算含量和RSD,RSD(n=6)分别为1.25%,表明样品制备方法重复性良好。试验结果见表3。
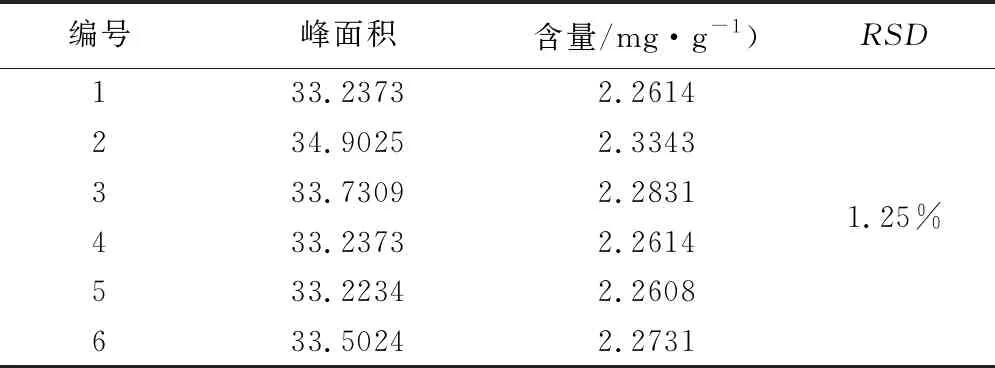
表3 盐酸水苏碱重复性试验结果表
2.2.6 准确性试验 计算回收率和RSD,样品中盐酸水苏碱平均回收率(n=6)为107.15%,RSD为1.90%。试验结果见表4。
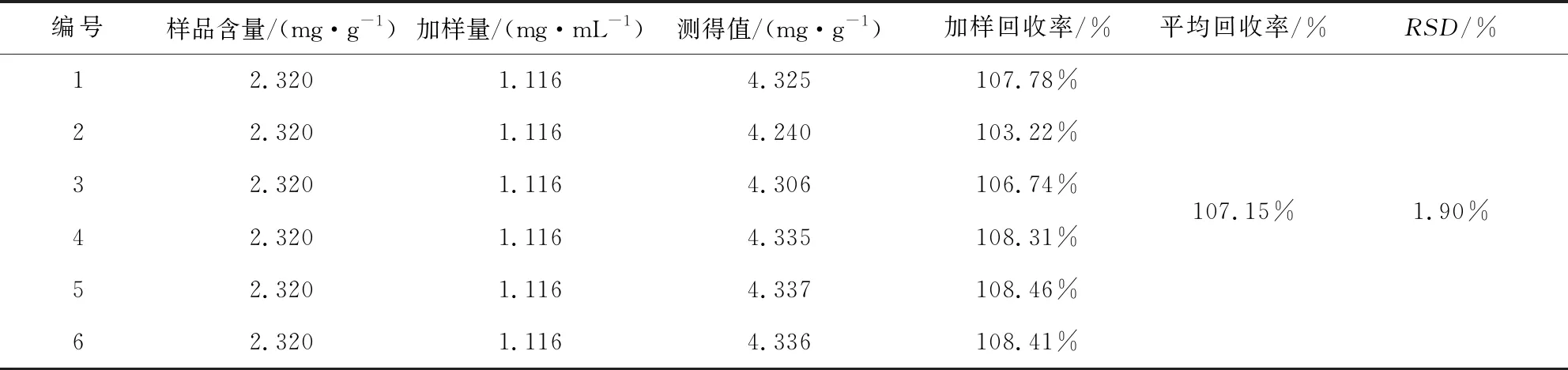
表4 盐酸水苏碱加样回收试验结果表
回收率计算公式为:
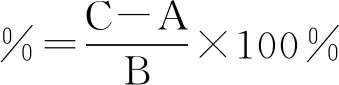
式中:A为样品所含被测成分量,B为加入对照品量,C为实测量。
2.2.7 样品测定试验 经测定,三批盐酸水苏碱的平均含量分别为2.320 、2.294、2.201 mg/mL,RSD=2.75%,说明不同批号的川芎益母散中盐酸水苏碱的含量稳定。
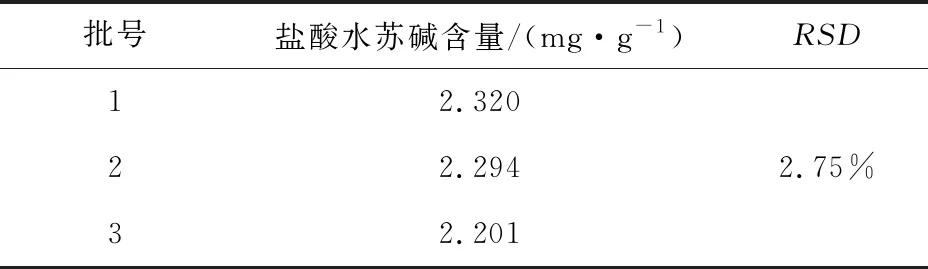
表5 样品测定盐酸水苏碱试验结果表
3 讨 论
益母草作为归芎益母散中的君药,其盐酸水苏碱含是衡量归芎益母散制剂质量的重要指标性成分,对盐酸水苏碱使用ELSD法进行定量研究,且结果表明此法测定归芎益母散中的盐酸水苏碱专属性强、稳定性好、精密度高,且阴性无干扰。对君药益母草和辅药川芎、当归和红花进行TCL研究,结果表明归芎益母散样品在与对照品相应的位置上,显示相同的颜色的主斑点,阴性样品无相应的斑点,TLC鉴别方法斑点清晰,专属性强,重现性好。因此通过对归芎益母散中益母草、川芎、当归和红花的定性鉴别及盐酸水苏碱的含量测定,能够有效控制归芎益母散质量。
在归芎益母散中益母草的TCL研究中,色谱方法参照2015版《中国药典》一部1006页[11],阴性对照样品与益母草对照药材色谱图中有相同颜色的斑点,但与对照品盐酸水苏碱无对应的斑点,参考药典中女金丸、产康复颗粒、四物益母丸等制剂的质量标准中益母草的鉴别[12],仅保留盐酸水苏碱对照品作为鉴别对照。
在红花TLC 鉴别方法中,将归芎益母散样品及对照药材红花提取液蒸干时,使用减压浓缩法至干,不可水浴蒸干。
在测定归芎益母散中盐酸水苏碱含量时,制备供试品过程中:加20 mL2.5%硫氰酸铬铵溶液后,冰浴(4 ℃)时间越久,测定含量呈上升趋势,3 h为最佳,因此冰浴时间为3 h。在加1%氯化钡溶液前浓缩为1 mL与不浓缩均对测定盐酸水苏碱含量结果无明显差异,因此在实验中在加1%氯化钡溶液前不浓缩。
4 结 论
研究建立的君药益母草、辅药川芎、当归和红花进行TLC鉴别阴性对照无干扰、斑点清晰且专属性较强。益母草中盐酸水苏碱的含量测定方法简便易行,重现性较好,为控制和评价归芎益母散的质量提供科学依据。
