QuEChERS-超高效液相色谱-串联质谱法检测牛奶中11种激素类药物残留
2021-09-16李永琴卜宁霞
陈 娟,李永琴,卜宁霞,马 岩,杨 奇
(宁夏兽药饲料监察所,宁夏银川,750011)
激素类药物对动物生理过程起着重要调节作用,可用于治疗动物疾病,促进动物生长发育、提高产量、增加蛋白沉积等[1]。奶牛在哺乳期会分泌一定量的激素,但养殖者为了加速奶牛发育和延长哺乳期会人为加入外源激素,并通过血液循环在牛乳中积累[2],牛乳中蓄积的激素药物会对人体健康造成较大危害,如增加生殖系统癌症风险[3-4]、诱发免疫性疾病[5]和影响儿童生长发育等[6],因此欧盟禁止在动物生产中使用激素药物[7],我国也在《食品安全国家标准动物性食品中兽药最高残留限量》(GB 31650-2019)和农业农村部第250号公告中分别对限用和禁用的激素类药物做出了相应规定[8-9]。
目前,国内外激素类药物的检测方法主要有高效液相色谱法[10]、气相色谱-质谱联用法(GC-MS)[11-12]、液相色谱-质谱联用法(HPLC-MS)[13-15]等,HPLC-MS具有样品前处理简单、灵敏度高、特异性强等优势,已经广泛应用于激素类痕量药物残留测定。但针对牛奶中激素检测方法的相关研究较少,且检测药物种类少、覆盖面窄,Liu Kun[10]等建立了牛奶中5种激素的自动在线固相萃取-高效液相色谱联用的检测方法,尤亮亮[13]等利用固相萃取-高效液相色谱串联质谱法测定了牛奶中9种性激素残留。同时《食品安全国家标准 动物源食品中激素多残留检测方法液相色谱-质谱法》(GB/T 21981-2008)中对激素残留测定的前处理方法繁琐、费时、成本高[16],不能够满足快节奏、大批量的检测需求。
QuEChERS法是一种快速、简易、廉价、有效、稳定、安全的前处理方法,现已广泛应用于兽药残留检测领域[17]。本研究利用QuEChERS法,通过优化前处理方法,结合超高效液相色谱-串联质谱技术,实现对群勃龙、诺龙、甲基睾酮、睾酮、黄体酮、丙酸诺龙、苯丙酸诺龙、勃地龙、司坦唑醇、美雄酮、丙酸睾酮共11种药物快速、高效的定性定量测定,可为牛奶中激素类药物监控提供技术支撑,保障牛奶质量安全。
1 材料与方法
1.1 仪器 Waters ACQUITY UPLC-Xevo TQ-s micro超高效液相色谱-串联质谱联用仪(美国Waters公司);AE-205电子天平(瑞士Mettler Toledo公司);CT15RT高速冷冻离心机(日本HITACHI公司);氮吹仪(美国Organomation Associates公司);Milli-Q integra 5超纯水仪(美国Millipore公司);多管涡旋振荡器(安简(北京)科技有限公司)。
1.2 试剂与材料
1.2.1 标准品 群勃龙、诺龙、甲基睾酮、睾酮、黄体酮、丙酸诺龙、苯丙酸诺龙、勃地龙、司坦唑醇、美雄酮、丙酸睾酮药物标照品(纯度≥95 %,除苯丙酸诺龙来源于中国食品药品检定研究院、美雄酮来源于上海安谱公司外,其他标准品均来源于德国Dr.Ehrenstorfer公司)。
1.2.2 材料 Cleaner MAS-Q 净化管(天津博纳艾杰尔有限公司);乙腈(色谱纯,美国Fisher公司);乙酸乙酯(分析纯,天津科密欧化学试剂有限公司);甲酸(质谱纯,美国Fisher公司);所用水为超纯水。
1.3 UPLC-MS/MS分析条件
1.3.1 色谱条件 色谱柱为美国 Waters BEH C18(50 mm×2.1 mm,1.7 μm);流动相A相为乙腈,B相为0.1 %甲酸水溶液;流速为0.3 mL/min;柱温为40 ℃;进样量为2 μL。流动相梯度见表1。
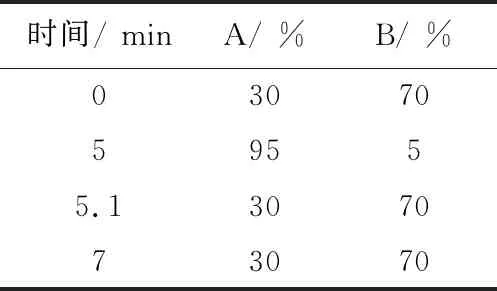
表1 流动相梯度洗脱条件
1.3.2 质谱条件 离子源:电喷雾离子源;扫描方式:正离子扫描;检测方式:多反应监测;毛细管电压:1.0 kV;源温:150 ℃;脱溶剂气温度:500 ℃;雾化气流速:1000 L/h;锥孔气流速:50 L/h。11种激素类药物定性、定量离子对及对应的锥孔电压和碰撞能量见表2。
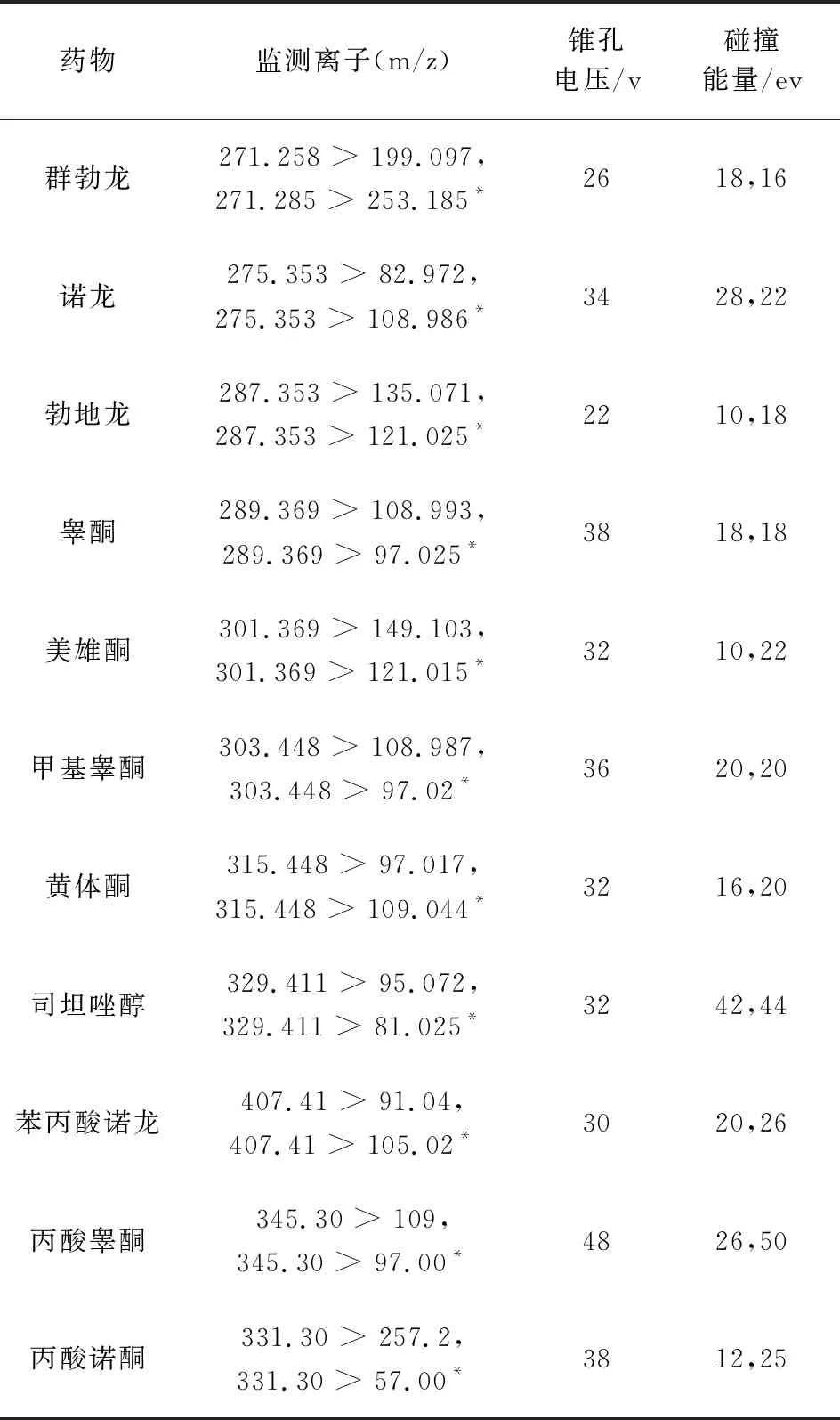
表2 11种待测药物质谱条件
1.4 实验方法
1.4.1 样品提取 准确称取混匀后的牛奶样品(2±0.05) g,置于50 mL塑料离心管内,加入乙酸乙酯10 mL,旋涡提取5 min,10000 r/min离心5 min,取上清液于15 mL具塞玻璃试管中,45 ℃氮气吹至近干。残留物用2 mL0.1 %甲酸乙腈水溶液(乙腈∶水=1∶1)溶解。
1.4.2 样品净化 取上清液1.5 mL至Cleanert MAS-Q净化管中,涡旋振荡1 min,10000 r/ min离心5 min,取上清液过微孔滤膜后,供液相色谱-串联质谱仪测定。
1.4.3 标准溶液的配制 标准储备液配制:分别精密称取11种激素类药物对照品各10 mg,分别置于10 mL容量瓶中,用甲醇溶解并稀释至刻度,配置成浓度为1 mg/mL的标准储备液,-20 ℃可保存6个月。标准工作液:分别准确吸取标准储备液适量,置于同一10 mL容量瓶中,用甲醇稀释成适宜浓度的混合标准工作液。
1.4.4 标准曲线绘制 准确吸取混合标准工作液适量,用空白样品提取液配成浓度为0.5、1.0、2.0、5.0、10、50、100 ng/mL的基质匹配混合标准工作溶液,从低浓度到高浓度测定,每一浓度进样3针,以特征离子质量色谱峰面积为纵坐标,标准溶液浓度为横坐标,绘制标准曲线。
2 结果与分析
2.1 线性关系 精密量取标准工作液适量,用空白基质稀释成含各药物浓度分别为0.5、1、2、5、10、50、100 ng/mL的系列混合标准工作液,充分混匀,过微孔滤膜,作为基质匹配标准溶液上机测定。以特征离子质量色谱峰面积为纵坐标(y),基质匹配标准溶液浓度为横坐标(x),绘制标准曲线,拟合回归方程。图1为0.5~100 ng/mL基质匹配标准溶液标准曲线图,11种激素类药物的回归方程及相关系数r见表3,由表3可以看出,11种药物在0.5~100 ng/mL的浓度范围内呈现良好的线性关系,r均大于0.998。
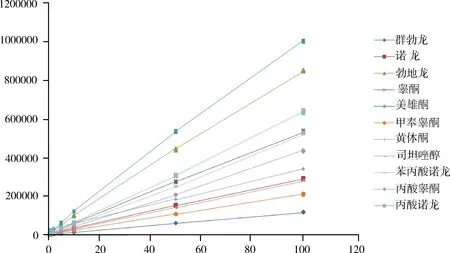
图1 基质匹配标准溶液标准曲线图(0.5~100 ng/mL)
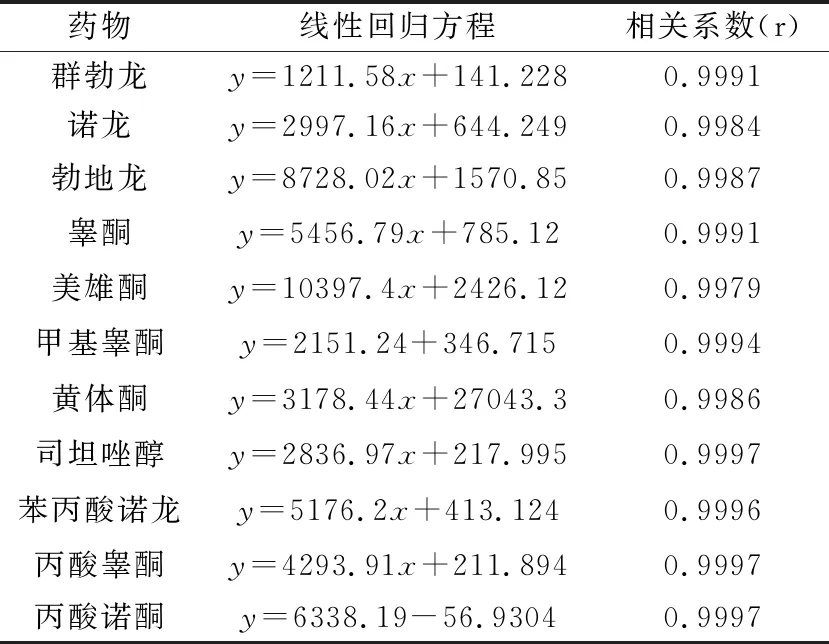
表3 11种激素类药物线性回归方程及相关系数
2.2 定量限 添加适量100 ng/mL混合标准溶液于2 g空白牛奶中,前处理后测定,依据各工作溶液对应峰的信噪比S/N>10(按PtP算),确定出激素类药物的定量限为1 μg/kg。
2.3 方法精密度与准确度 在空白牛奶中分别添加适量标准溶液,制成1、2、10 μg/kg三个不同浓度的空白添加试样,每批次同一浓度做5次平行试验,重复3次,进行回收率试验。试验结果见表4,由表4可以看出,空白牛奶中添加11种激素类药物添加回收率均在60.3 %~119.6 %;批内相对标准偏差在1.7 %~10.9 %;批间相对标准偏差在0.2 %~12.1 %,完全符合我国关于兽药残留检测试验技术规范的要求[23]。1 ng/mL空白牛奶基质匹配标准溶液中11种激素类药物特征离子质量色谱图见图2。
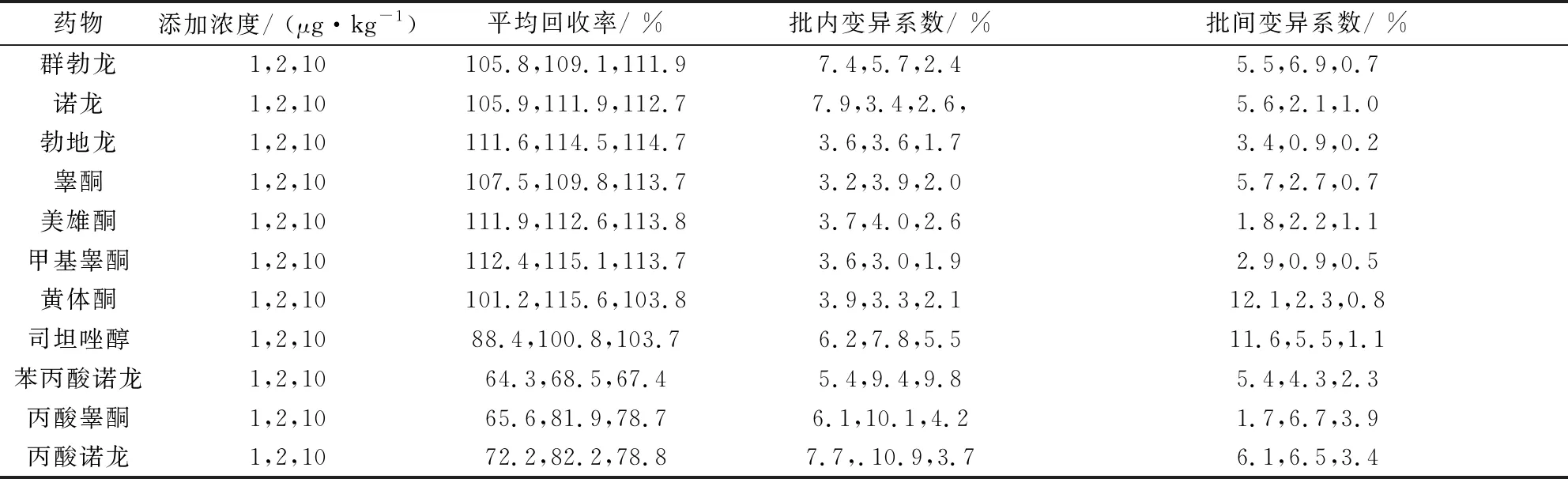
表4 11种激素类药物的加标回收率和精密度(n=5)
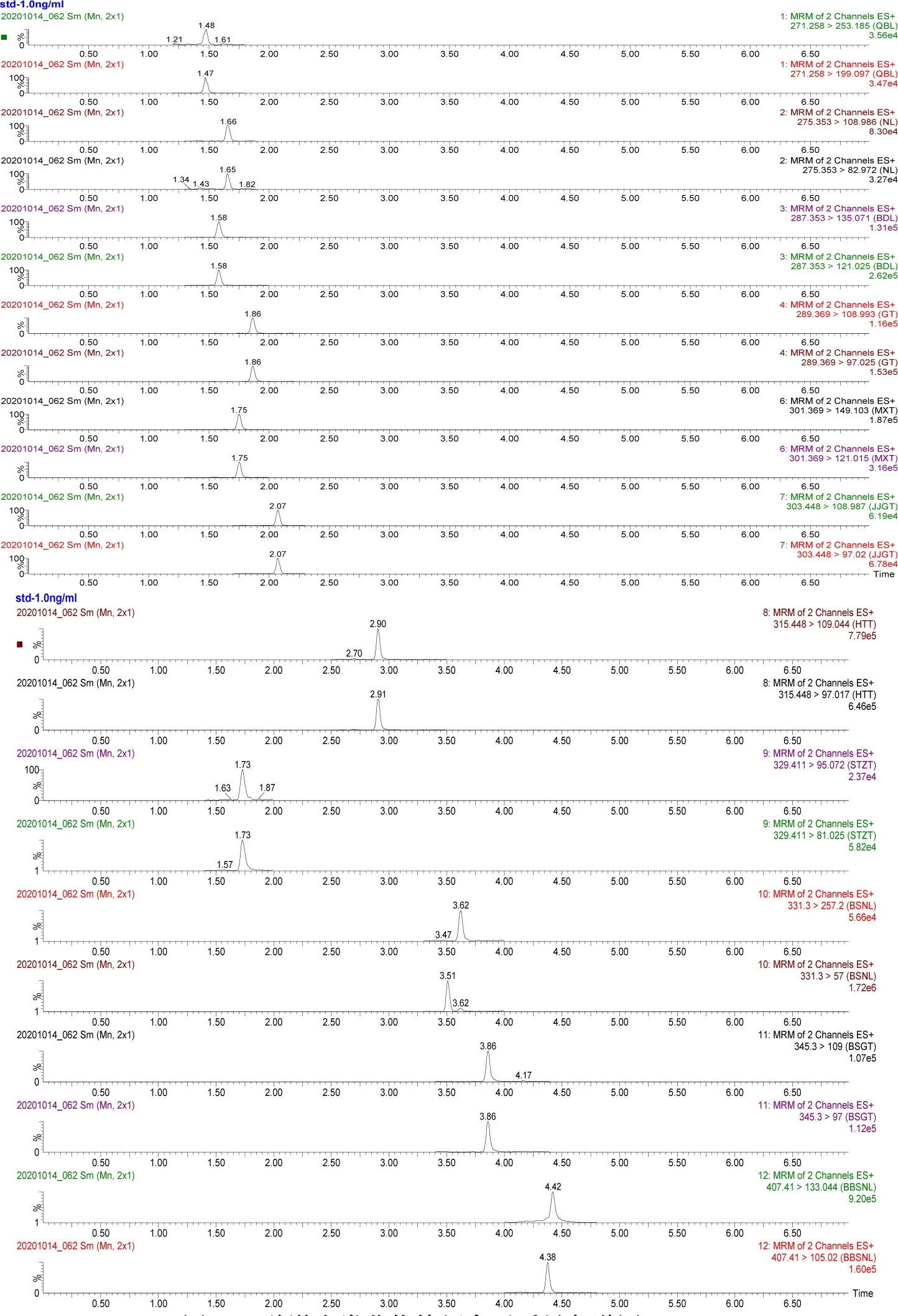
图2 11种激素类药物特征离子质量色谱图(1.0 ng/mL)
3 讨论与结论
3.1 质谱条件的优化 实验采用各药物0.2 μg/mL的标准溶液分别在电喷雾离子源正、负离子模式下,采用流动泵连续进样方式进行全扫描,在多反应监测模式下对质谱条件进行优化。结果发现:这11种激素类药物均在正离子模式下响应强度大,在此基础上选择丰度较强、干扰最少的两个子离子作为定性离子,其中丰度最强的子离子作为定量离子,同时确定出最佳碰撞能量和锥孔电压,具体质谱采集参数见表2。
3.2 提取溶剂的确定 目前,国内外激素类药物残留检测常用的提取溶剂主要为乙腈[10、18]、叔丁基甲醚[19]、乙酸乙酯[20]等。因此,实验采用空白添加法,分别考察了乙酸乙酯、乙腈、1 %甲酸乙腈溶液和叔丁基甲醚作为提取溶剂的提取效率。结果表明:叔丁基甲醚提取效率较低,乙酸乙酯和乙腈提取效率较高,但乙腈易使牛奶中的蛋白质变性、乳化,影响提取效果[21],且作为提取剂也会将牛奶中的水分也提出,降低氮吹浓缩速度,因此选择乙酸乙酯为提取溶剂。
3.3 净化方法的确定 QuEChERS净化方式所使用的吸附剂可将样品中的杂质、有机成分通过吸附、净化作用除去,减小样品基质对提取液中目标化合物的干扰,尤其是高蛋白、高脂肪的牛奶样品。实验采用HLB柱(500 mg/6 mL)、HLB柱(60 mg/3 mL)和Cleaner MAS-Q净化管分别对样品进行净化处理,考察样品净化效果和目标药物回收率。结果发现,三种方法均能起到较好的净化效果,但采用两种HLB柱净化后,丙酸睾酮、丙酸诺龙、苯丙酸诺龙和司坦唑醇4种药物回收率较低(仅为20 %~50 %),而采用Cleaner MAS-Q净化管净化后,11种药物回收率在60 %~120 %,这可能是因为Cleaner MAS-Q净化管中的吸附剂填料较为丰富,主要有PSA、PC、C18和MgSO4,其中PSA对脂类和糖类的去除效果较好,C18可去除脂类等物质[22],因此,为保证净化效果和回收率,实验选择Cleaner MAS-Q净化管净化样品,同时此方法还可在净化后直接收集上清液,过滤膜后上机检测,节省传统方法中的固相萃取和浓缩步骤,具有快速、节约、简单的优势。
研究建立了QuEChERS前处理方法结合超高效液相色谱-串联质谱法测定牛奶中11种激素类药物残留的检测方法。样品用乙酸乙酯提取,离心后氮吹浓缩,经0.1 %甲酸50 %乙腈水溶液复溶后用Cleanert MAS-Q净化管净化,过微孔滤膜后供UPLC- MS/MS测定。该方法在优化前处理方式后具有快速、节约、简单的优势,并在0.5~100 ng/mL的浓度范围内呈现良好的线性关系,r均大于0.998。在1~10 μg/kg添加水平下,药物添加回收率均在60.3 %-119.6 %,批内相对标准偏差在1.7%~10.9 %,批间相对标准偏差在0.2 %~12.1 %,定量限为1 μg/kg。该方法简捷、准确、灵敏度高,可满足对牛奶中多种激素药物残留监测的需要。
