儿童臂丛神经炎的临床特点(附4例报告)
2021-09-16胡笑月汤继宏黄静
胡笑月,汤继宏,黄静
臂丛神经炎是一种神经系统疾病,其特征是突然出现的肩部或手臂区域的疼痛,感觉丧失继之而来的肌肉无力、萎缩[1]。早期研究[2]认为臂丛神经炎发病率为0.02‰~0.03‰,但近期一项单中心前瞻性研究[3]显示其发病率为1‰,考虑与临床医师对疾病认识的提高有关。本病在所有年龄段均有报道,但发病中位年龄为20~40岁[4],因此在儿童中并不多见。本病可以为特发性或遗传性,后者被称为遗传性神经痛性肌萎缩,为常染色体显性遗传,主要由SEPT9基因突变所致[5]。特发性臂丛神经炎是一种比较常见的类型,常与病前感染和疫苗接种有关,提示其为免疫介导疾病。早期的诊断、识别及免疫治疗对于预后尤为重要。传统观点认为,儿童臂丛神经炎预后良好、恢复快,但一直争议不断。最近一项研究[6]表明,儿童臂丛神经炎预后欠佳。本研究报道了4例发病时间较为集中的儿童臂丛神经炎患者,并随访观察1年,结合文献阐明该病的特征、治疗及预后,以期加强临床认识和诊治经验。
1 临床资料
1.1 一般资料 系2018年10月至11月苏州大学附属儿童医院收治的4例臂丛神经炎患儿,诊断均符合以下标准[3,6]。上肢急性发作的疼痛和(或)以下异常:(1)上肢无力、感觉缺陷或肩胛骨摆动检查结果有1项及以上异常;(2)EMG提示臂丛神经异常,同时排除由外伤、恶性肿瘤、糖尿病和放射治疗等继发的臂丛神经损伤。男性3例,女性1例;年龄1岁10个月至6岁;病程3~6 d。所有患儿均无家族史。研究方案由本院伦理委员会审查和批准。
1.2 临床特征 见表1。患儿病前均有呼吸道感染病史,急性起病。4例均无疼痛表现,以肢体无力起病。病变累及右侧3例,左侧1例,无双侧受累者。其中1例累及同侧下肢,另1例累及同侧颅神经,表现为面肌瘫痪和伸舌歪斜。肌肉受累以近端为主。例1、例2、例3起病1周内完善EMG,例4在起病4周左右完善EMG。所有患儿EMG检查均提示臂丛神经复合运动动作电位(CMAP)波幅降低,例4还伴有失神经改变。3例行腰穿CSF检查,其中例3 CSF白细胞数52个/μl,生化正常。4例患儿均行颈椎及肩部MRI检查,其中例2患侧臂丛神经较对侧增粗。
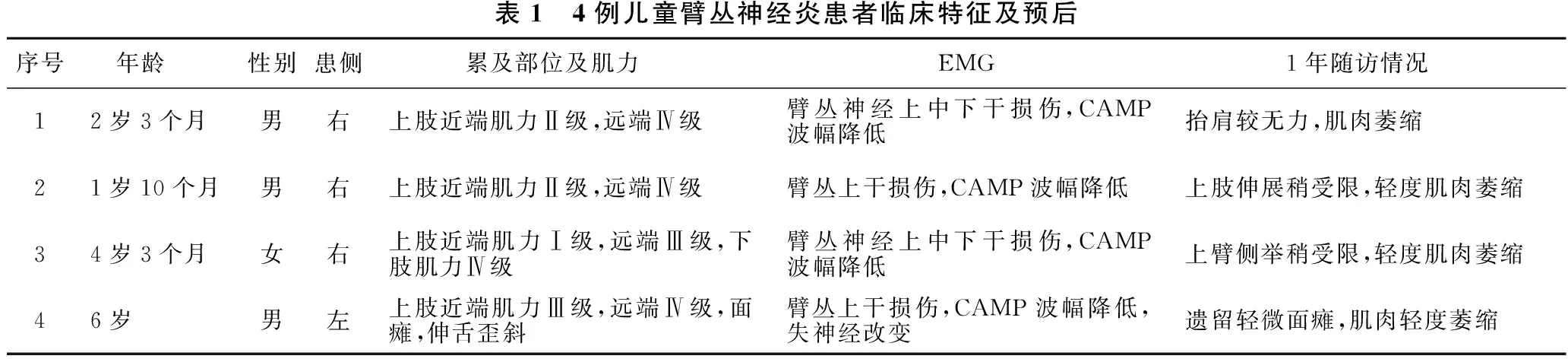
表1 4例儿童臂丛神经炎患者临床特征及预后序号 年龄性别患侧 累及部位及肌力 EMG 1年随访情况12岁3个月男右上肢近端肌力Ⅱ级,远端Ⅳ级臂丛神经上中下干损伤,CAMP波幅降低抬肩较无力,肌肉萎缩21岁10个月男右上肢近端肌力Ⅱ级,远端Ⅳ级臂丛上干损伤,CAMP波幅降低上肢伸展稍受限,轻度肌肉萎缩34岁3个月女右上肢近端肌力Ⅰ级,远端Ⅲ级,下肢肌力Ⅳ级臂丛神经上中下干损伤,CAMP波幅降低上臂侧举稍受限,轻度肌肉萎缩46岁男左上肢近端肌力Ⅲ级,远端Ⅳ级,面瘫,伸舌歪斜臂丛上干损伤,CAMP波幅降低,失神经改变遗留轻微面瘫,肌肉轻度萎缩
1.3 治疗与预后 例1、例2、例4均接受静脉甲泼尼龙[4 mg/(kg·d)]联合丙种球蛋白治疗,甲泼尼龙逐渐减量后改为泼尼松口服,2个月内泼尼松片逐渐减量。例3因家长拒绝糖皮质激素治疗,单用丙种球蛋白治疗。2个月后随访,所有患儿病情均有轻微改善,远端及下肢肌肉基本能正常活动,但近端运动仍受限。1年后随访,除例1抬肩较无力外,其余基本能正常活动,但均遗留不同程度的肌肉萎缩。
2 讨 论
臂丛神经炎在儿童中较少见,国内尚未见报道。1970年以前,由于小儿脊髓灰质炎暴发且与臂丛神经炎的临床表现相似,臂丛神经炎作为一种独立的临床疾病在儿童中还没有很好地被认识。一直到2000年,才有学者[4]系统总结了儿童臂丛神经炎的特征。据统计,有34%的患病儿童发病前有上呼吸道感染的病史[4]。在本研究中,4例患儿病前1~2周均有呼吸道感染,发病时间集中在2018年10月至11月的同一地区。鉴于儿童臂丛神经炎较少见,且本中心在一个集中的时间段内发现了4例患儿,考虑与某种未检测到的病原微生物流行和随后的免疫应答有关。Ooi等[7]曾报道了4例腺病毒流行感染后集中发病的臂丛神经炎患儿。
在成人臂丛神经炎中,肢体的疼痛和感觉受累是突出的特征,而这些在儿童,尤其在婴幼儿中通常是罕见或不明显的,因此没有疼痛并不能排除诊断[4]。在本组4例患儿中,没有患儿主诉有疼痛或查体时有疼痛表现,这使得诊断更加困难。但由于EMG和MRI的发展,误诊和诊断延迟的情况显著减少。
臂丛神经炎以上干受累多见,即上肢近端肌肉受损更明显,与本组病例报道相符。同例3和例4一样,部分患儿可以同时累及颅神经和腰骶丛神经,这类情况在有遗传因素的患者中更常见(55.8%)[8]。有文献[5,8]报道,EMG对臂丛神经炎具有诊断意义,神经病变形式以轴索损害为主,CMAP波幅降低。其中针极EMG异常率最高,一般在起病2周后出现失神经现象,所以本研究仅1例检查时间偏晚的患儿出现该现象。部分臂丛神经炎患者的臂丛MRI检查异常,主要表现为水肿、局灶性增厚和病灶的强化[4]。随着时间的推移,MRI的变化会随着临床症状的改善而消失。本研究只有1例患儿有臂丛神经影像改变,所以诊断更依赖EMG。
在所有年龄组患者中,男性的发病率均高于女性[9]。一般成人男女发病率比为2∶1,儿童为2.3∶1[4]。在本组中,比例为3∶1。这种性别分布与Guillain-Barré综合征(GBS)相似。尽管两者都是免疫相关的,但GBS一般更倾向于女性。基质金属蛋白酶9(MMP9)在GBS发病的性别差异中起重要作用[10],故不同性别的细胞因子表达和活化不同可能是男性更易患病的原因之一。
臂丛神经炎目前尚无特效治疗药物。考虑到该病可能是免疫介导的,故普遍采用免疫治疗,包括丙种球蛋白、糖皮质激素及血浆置换。研究[3]表明,发病后第1个月内用大剂量泼尼松治疗可以缩短疼痛的持续时间,并改善患者的功能。一些病例报道[11]也肯定了静脉注射丙种球蛋白的疗效。
在儿童患者中,免疫疗法的报道并不多见,疗效不一。臂丛神经炎本身具有一定的自限性,除遗传性的患者较易复发外,绝大多数患者在2年后可以恢复,但1年的恢复率仅36%[12]。Al-Ghamdi等[6]的研究纳入了22例臂丛神经炎的患者,平均随访1年,39%的患者遗留运动障碍。本研究4例发病1个月以上接受免疫治疗的患儿中,2例口服糖皮质激素,2例进行了血浆置换,平均随访1年余,仅25%遗留运动障碍,但因为样本量少无法进行统计学分析。Yamada等[13]报道了2例早期采用大剂量甲泼尼龙静脉冲击治疗的患儿,其中1例发病2个月时即完全恢复,另1例在6个月随访时病情改善,但未完全恢复。Al Masri等[14]报道了1例肾脏移植后应用他克莫司继发的臂丛神经炎患者,予静脉丙种球蛋白和大剂量甲泼尼龙冲击后未改善,后将他克莫司停用,替换为伊维莫司后患者完全恢复。提示当臂丛神经炎继发于药物损伤时,与免疫治疗相比,及时停用相关药物更为重要。本研究中,4例患儿在病程较早期都接受了积极的免疫治疗,随访1年基本可以正常生活,但仍有轻微运动障碍和肌肉萎缩。值得注意的是,4例患儿的症状在1年的长期随访中较2个月的短期随访有明显改善,说明预后与时间密切相关。
综上,本文报告了4例小儿臂丛神经炎患者,其临床特点与以往研究相似,EMG检查均提示臂丛神经受损。患儿在病程早期均接受免疫治疗,后期予康复训练,随访1年,预后尚可。但目前病例数太少,故仍需要多中心研究和更长期的随访来明确疾病预后。
