良性家族性婴儿癫痫的临床特点
2021-09-16赵静王训程楠杨任民韩丽孙道银年娜
赵静,王训,程楠,杨任民,韩丽,孙道银,年娜
良性家族性婴儿癫痫(BFIE)是一种少见的常染色体显性遗传的婴儿癫痫综合征,具有高度的遗传异质性。起病年龄3~24个月,高峰年龄4~7个月,多在1岁内起病[1]。本病发作形式多样,难以与其他婴儿期癫痫综合征相鉴别。但本病多呈良性病程,预后良好,多种抗癫痫药治疗均有效,故早期诊断治疗对患儿及其家庭尤为重要。现报道1例PRRT2基因突变和双侧额区放电的BFIE患儿,并结合文献进行分析。
1 临床资料
1.1 病例 患儿,男性,6.5个月。因“反复全身抽搐、双目上翻1个月”于2020年7月9日就诊于我院。患儿于5个月20 d玩耍时突然出现双目上翻,口唇吮吸样动作,双手攥拳,全身抽搐,持续1 min左右缓解,3~5 d发作一次。外院行EEG、MRI检查均无异常,诊断为“癫痫”,予以左乙拉西坦口服液2 ml 2次/d。家属述患儿服药后发作增多,几乎每天1~2次发作,此次为进一步治疗入住我院。个人史:G2P2,足月剖宫产,出生顺利,母妊娠期体健,无感染发热史。发育史:3个月会抬头,4个月会笑,认识妈妈。现能扶坐。家族史:父母体健,非近亲结婚。其父自述学龄期跑步时曾出现突然蹲下,双手痉挛,不伴有意识丧失,每次发作持续10 s左右,未治疗,30岁以后症状完全消失。胞姐12岁,生长发育正常。神经系统查体未见明显异常。实验室检查:血常规、生化、C反应蛋白正常,EB病毒、巨细胞病毒检查阴性。ECG、超声心动检查、头颅MRI平扫+DWI未见明显异常。EEG示婴儿异常EEG,睡眠期双侧额区成串低-高幅4~7 Hz尖波、欠典型尖慢波同步出现,偶见中-长程中-极高幅4~5 Hz尖化的θ波阵发,其间欠典型棘慢波夹杂(图1A、B)。基因检测(合肥金域医学检验实验室):患儿存在PRRT2基因16p11.2 NM_145239.2 Exon2 c.649dupC p.(Arg217fs)杂合变异,且其父亲为杂合携带者。一代测序图见图2。根据患儿病史特点[①5个月起病;②临床表现为无诱因的局灶性继发全面性发作,每日发作数次;③EEG发作间歇期双侧额区为主的局灶性痫样放电;④头颅影像学检查无异常;⑤排除低血钙、低血糖等代谢紊乱导致的惊厥;⑥起病前后智力、运动发育正常;⑦家族史:父亲有可疑的阵发性运动诱发的运动障碍(PKD)病史;⑧发现致病基因:患者及其父亲均存在PRRT2基因杂合突变],诊断为BFIE。予以奥卡西平口服液7.5 ml 2次/d。治疗7个月后回访,患儿自服用奥卡西平后未再发作,复查EEG(2021-02)示双侧额区放电仍存在,且数量未见减少(图1C)。

图1 EEG检查 A:睡眠期双侧额区成串低-高幅4~7 Hz尖波、欠典型棘慢波同步出现;B:中-长程中-极高幅4~5 Hz尖化的θ波阵发,箭头处为欠典型棘慢波夹杂;C:治疗7个月后复查示睡眠中双侧额区仍可见成串尖波,右侧明显

图2 基因检测结果
1.2 文献复习
1.2.1 检索策略 通过主题检索词“婴儿良性癫痫”及二级检索词“PRRT2基因”或“EEG”在中文期刊数据库(万方、维普、中国知网)检索到相关文献21篇,在英文期刊数据库(PubMed、Wiley)中检索到相关文献18篇。纳入及排除标准[2-3]:(1)起病年龄在1岁前;(2)无诱因的局灶性发作或局灶性发作继发全面性发作,多有丛集性发作(即24 h内发作≥2次)的特点;(3)EEG背景正常,发作期可为局灶起源的痫样放电,发作间期无典型痫样放电;(4)头颅影像学检查无异常;(5)排除低血钙、低血糖等代谢紊乱导致的惊厥;(6)起病前后智力运动发育正常;(7)有良性新生儿或婴儿癫痫家族史;(8)发作呈自限性或对抗癫痫药物反应好,且多数在2岁前癫痫发作缓解。对初筛后的文献摘要和内容进行阅读,剔除不合格的文献,获得符合要求的文献28篇,最终纳入5篇以BFIE报道为主的文献[4-8]。
1.2.2 一般资料 包括本文报道的病例,共纳入102个家系,331例患者(表1),均呈显性遗传。男197例,女134例;发病年龄2~13个月,其中5~7个月为发病高峰(282例,85.20%)。
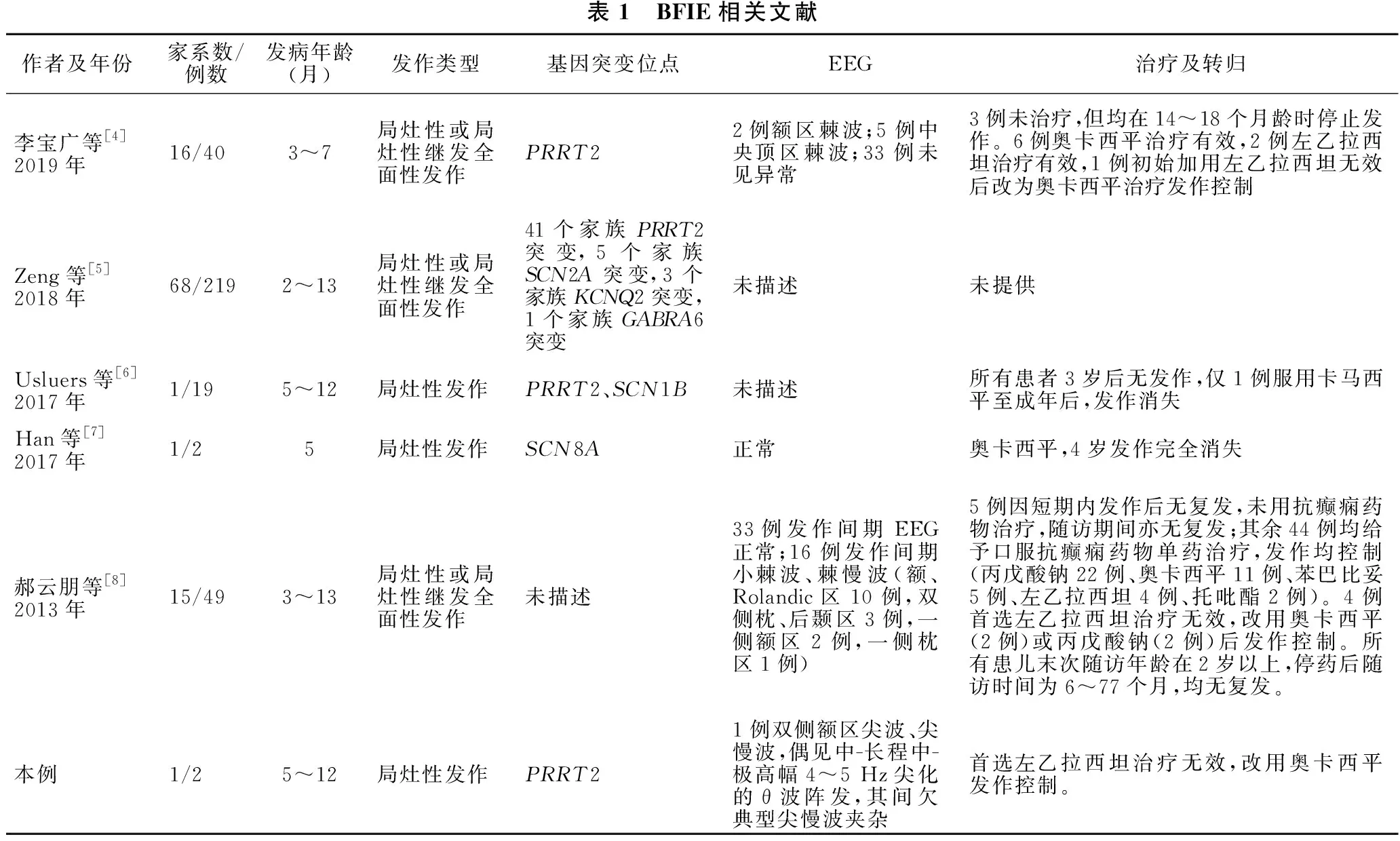
表1 BFIE相关文献作者及年份家系数/例数发病年龄(月)发作类型基因突变位点EEG治疗及转归李宝广等[4]2019年16/403~7局灶性或局灶性继发全面性发作PRRT22例额区棘波;5例中央顶区棘波;33例未见异常3例未治疗,但均在14~18个月龄时停止发作。6例奥卡西平治疗有效,2例左乙拉西坦治疗有效,1例初始加用左乙拉西坦无效后改为奥卡西平治疗发作控制Zeng等[5] 2018年68/2192~13局灶性或局灶性继发全面性发作41个家族PRRT2突变,5个家族SCN2A突变,3个家族KCNQ2突变,1个家族GABRA6突变未描述未提供Usluers等[6]2017年1/195~12局灶性发作PRRT2、SCN1B未描述所有患者3岁后无发作,仅1例服用卡马西平至成年后,发作消失Han等[7]2017年1/25局灶性发作SCN8A正常奥卡西平,4岁发作完全消失郝云朋等[8]2013年15/493~13局灶性或局灶性继发全面性发作未描述33例发作间期EEG正常;16例发作间期小棘波、棘慢波(额、Rolandic区10例,双侧枕、后颞区3例,一侧额区2例,一侧枕区1例)5例因短期内发作后无复发,未用抗癫痫药物治疗,随访期间亦无复发;其余44例均给予口服抗癫痫药物单药治疗,发作均控制(丙戊酸钠22例、奥卡西平11例、苯巴比妥5例、左乙拉西坦4例、托吡酯2例)。4例首选左乙拉西坦治疗无效,改用奥卡西平(2例)或丙戊酸钠(2例)后发作控制。所有患儿末次随访年龄在2岁以上,停药后随访时间为6~77个月,均无复发。本例1/25~12局灶性发作PRRT21例双侧额区尖波、尖慢波,偶见中-长程中-极高幅4~5 Hz尖化的θ波阵发,其间欠典型尖慢波夹杂首选左乙拉西坦治疗无效,改用奥卡西平发作控制。
1.2.3 临床表现 患者发作形式均为局灶性发作或局灶性继发全面性发作,常见发作表现包括反应减慢、双眼凝视、面色发绀。
1.2.4 EEG检查 多数患者发作间期EEG正常;24例发作间期异常放电,包括额区15例,中央区5例,枕区及后颞区3例,单纯枕区1例。
1.2.5 基因检测 87个家系进行了基因检测,共69个家系检出基因突变,突变率79.31%。其中59个家系(85.51%)为PRRT2基因突变,其中1个家系携带PRRT2、SCN1B两种突变基因;5个家系SCN2A突变;3个家系KCNQ2突变;1个家系GABRA6突变;1个家系SCN8A突变。
2 讨 论
既往研究[2]显示,出生后第1年的癫痫发病率为1.24‰,其发病率高于儿童期。根据国际抗癫痫联盟(ILAE)癫痫分类标准(修正版),其中58%可被归类为某种癫痫综合征。最常见的癫痫综合征为West综合征(0.41‰)和BEIF或非家族性婴儿癫痫(0.22‰)[9]。一般认为,新生儿、婴儿期发病的癫痫综合征常伴脑先天发育异常、不同程度脑损伤或代谢紊乱,发病年龄越小,预后越差。根据以往医院类机构的报道[10],仅21%~38%的患儿能达到正常发育水平。但近年临床观察[11]发现,一些婴儿期起病的癫痫,如West综合征的一种特发性亚型、良性婴儿癫痫、BFIE患者预后良好。早期诊断、恰当治疗可有效改善患儿预后,减少不必要的复杂检查和过多的药物治疗,减轻患儿家长的精神和经济负担。
根据发病年龄,良性家族性癫痫可以分为良性家族性新生儿癫痫(BFNE)、良性家族性新生儿-婴儿癫痫(BFNIE)及BFIE。BFNE多于出生后5 d起病[12];BFNIE多在生后2 d~3.5个月起病[13];BFIE高峰起病年龄为4~7个月[1]。在儿童期或青少年期,BFIE家系中的部分成员可出现PKD,这种BFIE的临床亚型被称为婴儿惊厥伴阵发性舞蹈手足徐动症(ICCA)[14]。本文共纳入102个家系的331例患者,无性别差异,呈显性遗传。发病年龄2~13个月,其中5~7个月为发病高峰,共282例,占患病总人数的85.20%,与既往国外文献报道[1]一致。本病实验室及生化检查无特异性,发作形式均为局灶性发作或局灶性继发全面性发作。发作期患儿临床表现缺乏特异性,家属常描述为反应减慢、双眼凝视、面色发绀、全身抽搐等全面性发作表现,这可能与患儿发作初期表现轻微、短暂,家长未关注到或局灶性发作很快泛化为全面性发作才被家长察觉到有关,因此仅凭病史很难对发作性质作出正确判断。EEG可以反映患儿颅内放电情况,对癫痫的发作定性具有重要作用。本研究多数患儿发作间期EEG正常,24例间隙期发现放电(15例额区异常放电,5例中央区异常放电,3例枕区及后颞区异常放电,1例单纯枕区异常放电),提示BFIE患者EEG虽无特异性,但间歇期局灶性放电部位多见于额区。Vigevano[1]曾报道在丛集性发作过程中的间歇期EEG可见单侧性慢波、枕区棘波等。本例患者发作间歇期双侧额区异常放电,偶见全导尖化的θ波阵发支持此观点,也证实了本病为局灶性癫痫的本质。目前对于治疗后EEG尚无相关报道。本例治疗7个月后复查,虽然无临床发作,但EEG仍可见睡眠期双侧额区尖波,数量未明显减少,且出现部位以右侧额区为著。这提示本病EEG改善可能要明显滞后于临床,本研究小组也将继续随访及收集此类病例加以证实。
既往国内外学者对BFIE遗传学及临床特征进行了研究。1992年Vigevano等[2]首次报道了本病。近年来,随着分子生物学技术的发展,有学者[5]发现BFIE呈常染色体显性遗传伴外显率不全,外显率为70%~90%,具有高度异质性。目前文献[15]报道的与本病相关的致病基因包括SCN2A、KCNQ2、SCN8A、ATP1A2、PRRT2基因。PRRT2基因被认为是本病的主要致病基因;KCNQ2是BFNE的主要致病基因;KCNQ2和SCN2A是BFNIE的主要致病基因[16-17]。PRRT2基因编码富脯氨酸跨膜蛋白2,编码340个氨基酸,产物为富含脯氧酸的跨膜蛋白,其功能尚不清楚。PRRT2基因是发作性运动诱发肌张力障碍、BFIE和ICCA家系的致病基因,并可导致其他多种良性发作性疾病[18-19]。目前很多研究[20-22]提示,PRRT2基因突变可导致多种神经系统发作性疾病,共同表现为婴儿、儿童期发病,呈发作性、良性病程,抗癫痫药物如卡马西平治疗有效等。由此,基于PRRT2基因多态性,将其导致的疾病称为PRRT2相关发作性疾病,包括发作性劳累诱发性运动障碍、家族性偏瘫性偏头痛、发作性共济失调、热性惊厥、发作性非运动诱发性运动障碍等。本研究纳入的102个家系中,87个家系进行了基因检测,69个家系出现基因突变,突变率79.31%。PRRT2基因突变占85.51%,SCN2A基因突变占7.6%,SCN1B基因突变占3.6%,KCNQ2基因突变占2.4%,GABRA6基因突变占0.6%,SCN8A基因突变占0.6%。这证实了本病遗传的高度异质性,也证实了PRRT2与本病高度相关,与既往文献[3,7,23-24]一致。有文献[4,25]报道,同为PRRT2基因突变,也有多种突变类型。少数为整体杂合缺失,多数为杂合突变(移码突变、无义突变、错义突变、终止密码子突变等)。本例突变位点为649dupC p.(Arg217fs),为移码突变,可导致所编码的蛋白质自217位氨基酸Arg开始发生移码,并导致翻译提前终止,是常见的致病变异,预计导致所编码的蛋白质发生截短,从而丧失正常功能。本例父亲携带相同致病基因,有婴儿期癫痫自愈病史,因此诊断明确。此外,结合患者父亲幼年时期PKD、成年后自愈的病史,高度怀疑本例家系为婴儿惊厥伴阵发性舞蹈手足徐动症家系。
本病呈良性病程,预后良好。原则上对于发作稀少和发作表现轻的病例可不用抗癫痫药物。有报道[26-27],本病对多种抗癫痫药物(如卡马西平,丙戊酸、苯巴比妥和唑尼沙胺良等)反应良好。但本病患儿本质上为局灶性起源,治疗应首选对局灶性发作有效的药物。本研究纳入的研究除文献[18]未描述治疗情况外,多数患者2岁后停止发作,所有患者临床发作均在4岁前完全消失。值得注意的是,本例患儿初始予以左乙拉西坦治疗,发作反而增多,改用奥卡西平后控制理想,2个月后随访发作完全消失,与李宝广等[4]的报道相似:在16个PRRT2突变家系中,3例初始加用左乙拉西坦的患者无效或控制不佳,改用奥卡西平后发作控制。这提示在选用左乙拉西坦治疗本病时应更谨慎。但因样本量不足,此观点还需更大样本进一步论证。
本研究通过本病例及相关文献分析,总结了BFIE的临床、EEG、致病基因特点。在临床上如有婴儿期起病的局灶性癫痫(如丛集性发作),且智力、运动发育正常,需考虑本病。
