“困难序列”多肽的合成方法
2021-09-15许小玲张荣江易小庆陈伟铭
许小玲,张荣江,谢 欣,3,易小庆,3,陈伟铭,3,张 剑,3,江 峰,3
(1.赣南医学院2019级硕士研究生;2.赣南医学院药学院;3.心脑血管疾病防治教育部重点实验室,江西 赣州 341000)
多肽是由多种氨基酸通过肽键结合而成的一种生物活性物质,具有广泛的生物活性及良好的安全性,广泛应用于医药、食品、化妆品、农业及畜牧业等领域[1]。作为化学合成多肽的重要途径,固相合成因避免了液相合成中冗长的重结晶或分离步骤,减少了合成生产过程中的损失而得到了广泛的应用[2]。但是,对于一些含有较多疏水性氨基酸片段且容易出现β-折叠等二级结构的多肽仍然难以合成,尤其是出现在第6~16个氨基酸残基的“困难序列”[3],几乎每一个氨基酸的脱保护和偶联都难以顺利完成,缺失肽和截头肽较多,收率很低。
目前解决多肽“困难序列”合成困难的方法有很多,包括使用混合溶剂、减少树脂担载量、使用高离液盐、片段缩合、引入化学修饰的肽链和采用微波技术合成等[4]。本文对“困难序列”形成的原因及其解决方法进行了综述,并总结了一些经典“困难序列”多肽固相合成的基本策略。
1 “困难序列”形成的主要原因
根据形成原因的不同,多肽“困难序列”可以分为随机和非随机两种类型[5],随机型“困难序列”与氨基酸疏水性及立体位阻有关。当氨基酸的立体位阻较大(如氨基酸的侧链具有较大保护基)时,酰化试剂扩散困难;或树脂的负载量较大时,树脂上的肽链溶剂化不完全,偶联结果都很差[5]。随机困难肽可以通过疏水性参数来粗略表示[6],现在仍没有一条“黄金规则”能准确预测。一般认为,当疏水性参数达到正数,其合成就相当困难[7]。非随机型“困难序列”是指易形成具有稳定且特异性β-折叠结构的肽链序列。该类序列的肽链在一定长度时很容易发生氢键缔合,且与树脂载体之间也容易出现分子聚集现象,多肽分子的氨基可能埋没在二级结构的深处,进而阻止缩合反应,此现象常出现在难溶多肽的合成中[4,8]。使用9-芴甲氧羰基(Fmoc)保护的氨基酸合成多肽时容易形成这种β-折叠结构,合成过程需要改变反应条件如提高温度等破坏这种构型,但这可能会导致产物消旋化或提前脱除Fmoc保护等不良反应[7]。
2 解决“困难序列”多肽合成困难的方法
解决“困难序列”多肽合成困难的方法有很多,可以从改变反应体系、改变反应条件、改变合成策略、采用新的合成技术手段等方面进行。
2.1 改变反应体系
2.1.1 使用混合溶剂多肽树脂在溶剂中达到充分溶胀状态时,有利于缩合反应的进行。当选用如N-甲基吡咯烷酮(NMP)、N,N-二甲基甲酰胺(DMF)、二甲亚砜(DMSO)、N,N-二甲基乙酰胺(DMAc)等受氢溶剂时,其羰基(C=O)与肽链上的N-H可以形成氢键,从而抑制了肽链自身形成氢键而造成的聚集,进而抑制β-折叠的生成。因此,选用合适的混合溶剂,比如DMSO/DMF、6N胍啶/DMF、异丙醇/DMF、二氯甲烷(DCM)/DMF/NMP、三氟乙醇(TFE)等有助于缩合反应的进行。黄惟德在《多肽合成》一书中提到,DMF为溶剂的反应液中加入20%体积的三氟乙醇(TFE)可帮助接肽速率提高[9]。朱亮亮等在大位阻氨基酸Fmoc-Arg(Pbf)-OH与Rink Amide-AM树脂的高效缩合中以DMAc/DCM为反应溶剂,连接率高达93%[10]。
2.1.2 使用溶胀性能更好的树脂当树脂的负载量较大时,树脂上的肽链溶剂化不完全,偶联结果很差[5]。使用溶胀性较好的树脂,同时减少树脂担载量,有利于“困难序列”多肽的合成。比如Winkler等在合成“困难序列”多肽K5(序列:CGGKVSALKEKVSALKEKVSALKEKVSALKEKVSA LKE)时,采用PEG-PS树脂代替常规的PS/DVB树脂,使得粗肽收率有了显著性提高[11]。然而,由于成本高昂,这种方法不适合于大规模的工业生产[12-13]。
2.1.3 选用高效缩合试剂选用高效缩合试剂,可以有效地促进缩合反应的顺利进行,也是解决“困难序列”多肽合成困难的一种常见方案,见图1。通常DIC的反应活性低于HBTU,HBTU的反应活性低于TBTU、PyBOP和HATU[14-15]。所以,在反应活性比较低的时候,可以选择高活性的缩合试剂。

图1 常见的高效偶联试剂
2.2 改变反应条件
2.2.1 提高反应温度提高反应温度有助于降低链聚合现象。BACSA B等[16]在合成1个九肽的实验中发现,将合成温度提高到60℃左右,最终产品的纯度可达到83%。然而,提高反应温度会导致新的不良反应的发生,比如侧链保护基的断裂[17]。所以,在尽可能减少不良反应的情况下,可以提高反应的温度。
2.2.2 调节反应p H值调节反应pH值能改变多肽在溶剂中的溶解度,从而提高“困难序列”多肽的合成效率。比如QIN X等[18]、AJIKUMAR PK等[19]研究了体系的pH值对多肽收率的影响,发现根据多肽序列中酸性和碱性氨基酸的数量,适当调节溶剂的pH值,可以提高肽链的溶解性,促进反应的进行。然而,由于在该pH值条件下,多肽保护基可能不稳定,所以不常使用[20]。
2.2.3 使用高离液盐高离液盐能破坏肽链间的氢键,从而破坏β-折叠结构,提高“困难序列”多肽的合成效率。常见的高离液盐有尿素、LiCl、NaClO4等。比如,WESTALL FC等[21]在合成“困难序列”多肽时,发现加入尿素之后可使Asn及Glu的缩合率由70%提高至100%。这可能是由于尿素具有受氢能力,干扰了肽链上的氢键缔合,从而促进了多肽的合成。然而,使用高离液盐会降低反应液的浓度,对反应速率有一定影响。
2.3 改变合成策略
2.3.1 片断合成法常规多肽固相合成,是从碳端开始按照氨基酸的顺序依次合成。当遇到“困难序列”时,采用片段合成法先制备带有保护基的中间序列片段,再经过进一步活化、缩合得到保护的多肽,最后脱去保护基得到目标多肽。片段合成要考虑多肽片段的长短、连接位点和固相载体这3个关键因素。由于肽段羧基端氨基酸比氨基端氨基酸更容易发生消旋反应,因此首先选择多肽片段C末端的氨基酸。适合做C末端的氨基酸有Gly(甘氨酸)、Pro(脯氨酸)、Glu(谷氨酸)、Leu(亮氨酸)和Asn(天冬酰胺)等[22]。周建华等[23]以王树脂为固相载体制得25-34肽树脂;以2-氯三苯甲基氯树脂为固相载体制得1-11、12-16和17-24等3个片段的全保护肽;然后将3个片段的全保护肽按照肽序依次缩合到25-34的肽树脂上,经三氟乙酸切割并脱除侧链保护基得特立帕肽粗品。SHIL等[24]采用片段合成法顺利地合成了阮病毒蛋白片段。张颖等[25]在合成含有3股β折叠的齐考诺肽(25肽,序列:CKGKG AKCSR LMYDC CTGSC RSGKC-NH2)的 过程中,采用固相合成得到4个片段,分别是[19-25]A、[12-18]B、[6-11]C和[1-5]D,再将B、C、D依次偶联到肽树脂A上,最后从树脂上裂解得到线形粗肽。该方法相对于传统的逐步缩合方法,缩短了合成周期,提高了粗肽的收率和纯度,为长肽的合成难题提供了可行性的解决方案。
2.3.2 引入化学修饰的肽链引入化学修饰的肽链,将“困难序列”多肽的高级结构暂时性破坏,促进肽的溶解,从而有利于多肽的合成。比如,当肽链中含有脯氨酸片段时,缩合形成的酰胺键没有可以形成氢键的质子,脯氨酸的α碳原子处在五元环的刚性结构中,从而抑制了β-折叠结构的形成,所以,在合成“困难序列”时可以考虑引入脯氨酸来减少合成的难度。WHITE P等[26]在合成一些较长多肽序列(95肽)的时候引入伪脯氨酸二肽结构(类似脯氨酸结构)保护氨基酸来改善聚集问题,见图2。Ser、Thr和Cys 3个氨基酸可以和序列中邻近的氨基酸形成一个伪脯氨酸二肽(fPro),肽链在树脂上组装完成后,放在TFA溶液中搅拌,将多肽从树脂上裂解下来,同时恢复正常的序列,不会影响多肽的性质[27-28]。
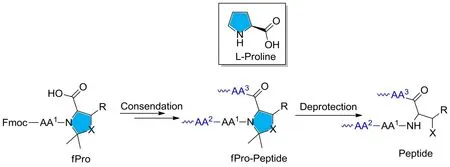
图2 伪脯氨酸(fPro)引入和脱除
2.3.3 氨基酸上引入取代基在氨基酸的氨基上引入不同取代基,可以改变合成过程中肽链的延伸方向,从而破坏肽链聚集现象,促进缩合反应的进行。TICKLER A K等在amyloid-β(含有40-43氨基酸残基)的固相合成中每6~7个氨基酸残基就引入1个2-羟基-4-甲氧基苄基(Hmb)保护基,肽链在树脂上组装完成后,将多肽从树脂上裂解下来,同时把Hmb取代基除掉得到产物(图3)[29]。OFFER等[30]引入Hmb类似物2-羟基-4-甲氧基-5-硝基苄基保护基,很好的提高了聚集肽等困难系列的合成效率。但是引入取代基需要对保护的氨基酸原料进行改进,操作不方便,成本很高。

图3 Hmb保护基的引入和脱除
2.4 采用新的合成技术手段微波合成、红外加热和超声辐射作为新的技术手段,表现出加热快速、均质与选择性好等特有优势,已被应用在“困难序列”多肽的合成中。HOJO K等[31]利用微波加热方法在水溶液中成功地合成了公认的“困难序列”酰基载体蛋白ACP(65-74)肽,克服了原有化学合成污染大、三废多的缺点,纯品收率达到38%。HOJOK等[32]证明微波辐射可以成功地提高亮氨酸-脑啡肽和聚集肽Val-Ala-Gly-OH的合成产率和纯度。MARO等研究了超声波对多肽固相合成的影响,发现超声辐射改善了具有“困难序列”的肽的合成,但并没有加剧主要的不良反应[33]。DERDA等提出了用红外加热的方式来促进“困难序列”的肽的合成[34]。利用这些新的技术手段合成多肽,能够极大地提高产物的产率及纯度,同时也为降低反应成本提供了可能性。
当上述单一方案不能达到目的时,WALJI等将提高反应温度、用微波或红外加热以及溶剂化聚乙二醇树脂联合使用,成功得到“困难序列”Omomyc[35]。
3 经典的“困难序列”多肽合成策略
3.1 胸腺法新的合成胸腺法新(胸腺肽α1)的序列为Ac-SDAAV-DTSSE-ITTKD-LKEKK-EVVEEAEN-OH,是一个28肽。主要用来治疗慢性乙型肝炎,具有良好的免疫调节作用[36]。胸腺法新的酸性残留位置、疏水性氨基酸的连续存在(Thr-Thr、Val-Val)和需要大量的保护基(20个侧链保护基)等造成了其固相合成的困难。采用Fmoc策略合成时,C端第1个氨基酸会用到Fmoc-Asn(Trt)-OH,立体位阻很大。连续疏水性缬氨酸(Val-Val)的出现,会造成β-折叠的生成;已有大量的实验证明,偶联至第9个氨基酸(K)时,多肽的纯度会急剧下降,胸腺法新是一个典型的“困难序列”多肽(图4)。

图4 胸腺法新的氨基酸序列
自胸腺法新被发现以来,多位研究者采用不同的策略致力于该“困难序列”的合成。最早来自于1980年美国罗氏公司和美国洛克菲勒大学的研究者,他们对主链氨基采用Boc保护的固相合成法,总收率不到7%[37],后经WANGTW等[38]的改进(采用羟甲基-苯乙酰胺甲基树脂,增强了肽-树脂键的稳定性),纯化收率提高到34%。然而,采用Boc保护基的固相合成法,要频繁用到TFA和HF等腐蚀性气体,污染大,肽树脂不稳定,如今已很少被采用。
为了提高胸腺法新的产率,1985年罗氏公司的FELIX A M等[39]尝试了片段合成法,然而,最后4步的收率仅为30%。1988年德国图宾根大学的ECHNER H等[40]报道了用高效偶联试剂Bop做活化剂,氨基采用Fmoc保护,侧链采用t-Bu或Boc保护的固相合成法,粗品收率达到76%。但经过纯化后,纯品收率比较低。
在这些解决方案中,最成功的莫过于刘标等,他们以HBTU/HOBt/DIPEA体系作为缩合试剂,在易形成α螺旋和β转角的肽序位置,通过采用HOBt/DIC补投的方法促使偶联反应完全。以茚三酮显色的方法对反应进行监控,获得的粗肽纯度为67.2%。以反相液相色谱法为主要分离手段,通过多步分离实现纯化。胸腺法新产品纯度达99.5%以上,总收率为17.2%[41]。另一个成功的解决方案是2009年西班牙生物医学研究所和瑞士Lonza公司合作提出的,他们采用改进的Fmoc/t-Bu的固相合成法,C端的Asp主链羧基用t-Bu保护,侧链羧基连接到PEG氨基树脂上,即采用Fmoc-Asp-OtBu作为第一个引入的保护氨基酸。该方法的粗品纯度达到90%[42]。
3.2 扶素康的合成扶素康是根据HIV膜蛋白gp41的结构设计的新一代膜融合抑制剂,是一个36肽,其序列为:SWETW-EKEIE-NYTKQ-IYKILEESQE-QQDKN-EKDLL-E[43]。然而,由于肽链中含有大量的疏水性氨基酸(W、T、I、L)和立体位阻很大的氨基酸(W、Q、N),以及需要大量的保护基(30个侧链保护基)等造成了其固相合成的困难。扶素康的早期合成采用固相逐步化学合成法。采用Fmoc保护策略、王树脂为固相载体、反应溶剂为DMF、HBTU/HOBt/DIPEA为缩合剂,保护氨基酸2倍过量进行缩合反应、TFA/苯酚/EDT/水为切割条件,得到的粗肽纯度和收率都很低[44]。
为了提高扶素康的产率,郭一琼等[44]采用片段合成的策略。他们以2-氯三苯甲基树脂和王树脂为固相载体,HBTU/HOBt/DIPEA为缩合剂,DMF为反应溶剂,用HOAc(乙酸)/TFE/DCM将全保护多肽片段由树脂上切割下来。将全保护多肽片段依次偶联,再用TFA溶液除去保护基。该条件下合成的扶素康粗品收率达67.31%,按标准曲线定量分析粗品纯度为38.75%[44]。
4 总结和展望
近年来,多肽固相合成技术发展迅速。但由于空间位阻等随机型和β-折叠结构的非随机型“困难序列”的存在,导致缩合反应纯度低、收率低。通过使用混合溶剂、高效率的活化剂和新型树脂等改变反应体系的方法,提高反应温度、调节反应体系的pH值和使用高离盐液等改变反应条件的方法,以及片段缩合、引入取代基等新型合成策略,为实现“困难序列”多肽的合成提供了解决思路。但是,有些策略仍只能处在实验室研究阶段,无法实现大规模生产。
随着多肽应用范围越来越广、研究越来越深入,固相合成多肽在未来依然有巨大的发展优势。随着高效率的活化剂和新型树脂的不断涌现,为解决“困难序列”多肽提供了新的可能。
