Beals-Hecht 综合征1 例报告
2021-09-13周焕珍王爱萍
周焕珍 王爱萍
昆明市第一人民医院儿童生长发育管理中心(云南昆明 650011)
Beals-Hecht 综合征(Beals-Hecht syndrome,BHS,MIM 121050)也称先天性挛缩蜘蛛样指畸形(congenital contractural arachnodactyly,CCA),是由于原纤维蛋白缺陷引起的一种常染色体显性遗传性结缔组织疾病[1]。文献报道原纤维蛋白-2(fibrillin-2,FBN2)基因为BHS的致病基因[2]。BHS的特征表现为多发性关节挛缩(尤其是肘部、膝盖和手指关节)、蜘蛛样指、严重脊柱后凸畸形、耳廓异常和肌肉发育不良。其症状和体征与马凡综合征(Marfan syndrome,MFS,MIM 154700)相似,但是BHS区别于MFS的显著特征是手指的屈曲畸形和外耳畸形。由于缺乏确切的临床诊断指标,此病的临床表型又与MFS有一定交叉,在没有基因诊断前BHS确诊较为困难。本文报告1例通过基因检测确诊的BHS患儿,并结合既往文献复习该病的病因、临床表型、诊断及治疗。
1 临床资料
先证者,男,3个月28天,因发现双上肢及双下肢屈曲挛缩3月余就诊。患儿系G1P1,36周剖宫产,出生体质量2.2 kg,身长46 cm。出生史无异常,否认窒息抢救史,否认重度黄疸史。生后母乳喂养,无喂养困难,无便秘及长期腹泻史。父母均为汉族,非近亲结婚,智力、面容及骨骼发育无异常;母亲双手指细长、无弯曲。体格检查:体温36.5℃,心率122次/min,呼吸28 次/min,体质量3.5 kg。一般情况及反应可,神清,前囟约1.5 cm×1.5 cm,平软,不能竖头,长头畸形,小下颌、高腭弓、短颈、皱耳(图1 A),颈抗(-),双眼无异常,无蓝色巩膜,双侧瞳孔等大等圆,对光反射存在,浅表淋巴结无肿大,全身无皮疹及色素沉着,甲状腺无肿大,呼吸平稳,双肺呼吸音粗,未闻及干湿啰音,心律齐,心音有力,心前区闻及二级收缩期杂音,腹软不胀,肝脾未触及,肠鸣音正常,四肢肢端暖,足跟毛细血管充盈时间1~2 s,双侧肘关节屈曲挛缩,双手指细长、屈指、拇指内收(图1 B),手指尺侧偏斜,双侧膝关节屈曲挛缩、下肢肌肉发育不良(图1C),双足趾细长、屈趾、马蹄内翻足(图1D),四肢肌力、肌张力无法评估,病理征(-),余神经系统无异常。实验室检查:血、尿、粪常规无异常,血生化、电解质、甲状旁腺素、甲状腺功能均无异常,血清25-羟维生素D 84.11 nmol/L,血气分析 pH 7.389,剩余碱-1,二氧化碳分压27 mmHg,血乳酸1.6 mmol/L。腹部及颅腔超声均无异常。心脏彩超示卵圆孔未闭,双上腔静脉。脑电图无异常。X 线片示右侧尺骨近端明显弯曲(图2 A),左侧肱骨中段陈旧性骨折后大量骨痂生长(图2B),双侧股骨、胫腓骨细长,骨髓腔略变窄、骨干略弯曲(图2 C);颈、胸、腰骶椎骨质未见明显异常,各椎体排列整齐,椎间隙未见明显狭窄。头颅磁共振(MRI)示双侧额、颞、顶叶白质区含量稍高,透明隔腔隙稍增宽。
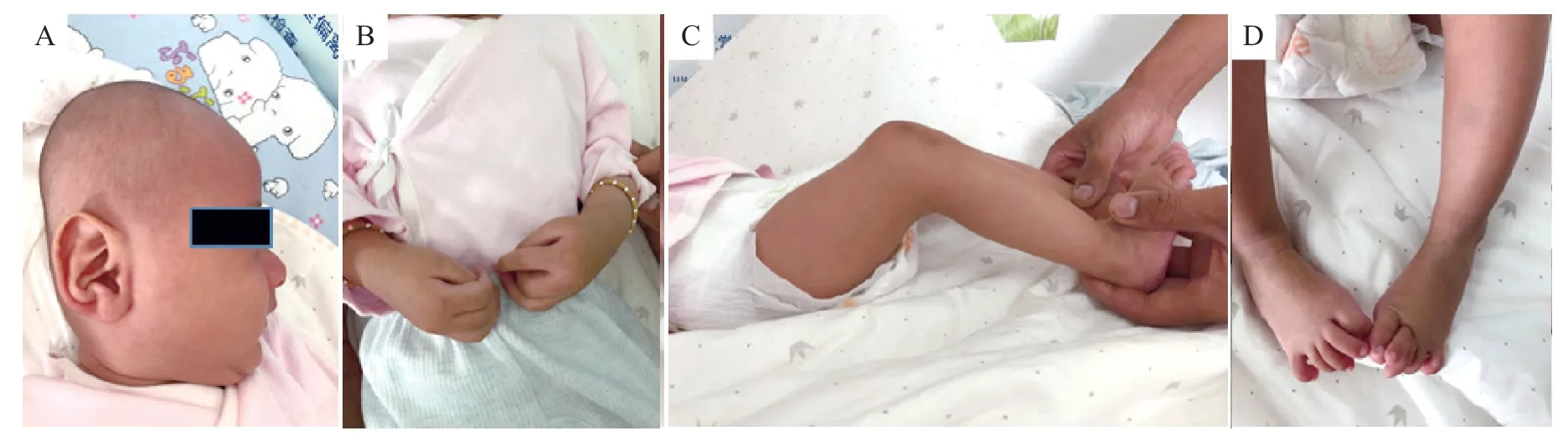
图1 患儿临床特征
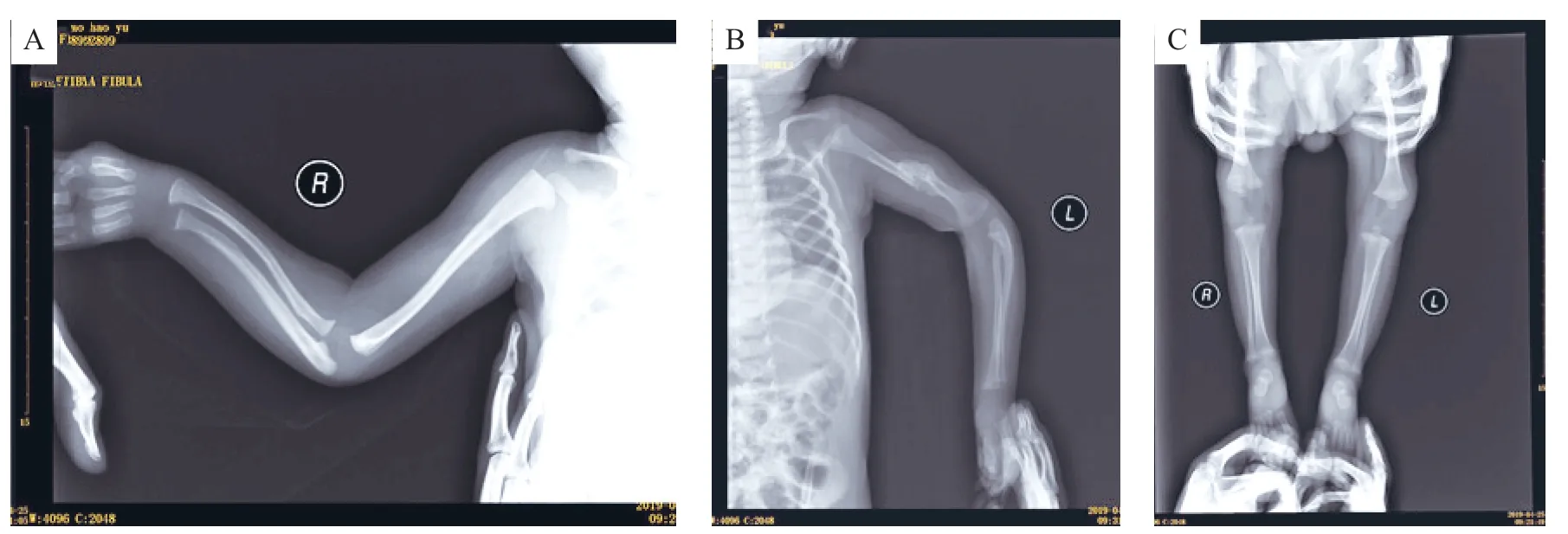
图2 患儿影像学表现
因患儿存在四肢关节挛缩畸形及皱耳,四肢X线片异常,考虑存在骨代谢性疾病或骨骼遗传性疾病,故行基因检测。经医学伦理审核及家长知情同意,留取患儿及父母外周血送北京迈基诺医学检验公司,行骨骼系统遗传病基因检测和分析panel-V2。结果显示,患儿FBN2基因有2个杂合变异c.2944T>G及c.6518 A>G。c.2944 T>G(编码区第2944 号核苷酸由胸腺嘧啶变异为鸟嘌呤),导致第982 号氨基酸由半胱氨酸变异为甘氨酸(p.C 982 G),为错义变异(图3)。根据美国医学遗传学和基因组学(American College of Medical Genetics,ACMG)指南[3]对变异进行分析:①PS 2,经家系验证分析,其父该位点无变异,其母为杂合变异;②PM 2,在正常人群数据库中为低频变异;③PP 3,生物信息学蛋白功能预测软件SIFT、PolyPhen_2、MutationTaster、GERP++、REVEL均预测为有害;④人类基因突变数据库(Human Gene Mutation Database,HGMD)未有该位点的相关性报道。c.6518A>G(编码区第6518号核苷酸由腺嘌呤变异为鸟嘌呤)导致第2173号氨基酸由天冬酰胺变异为丝氨酸(p.N2173S),为错义变异(图4)。根据ACMG指南对变异进行分析:①PS 2,家系验证分析显示其父该位点杂合变异,其母无变异;②PM 2,在正常人群数据库中的频率为0.00008;③PP3,生物信息学蛋白功能预测软件SIFT、PolyPhen_2、MutationTaster、GERP++、REVEL均预测为有害;④HGMD数据库未有该位点的相关性报道。
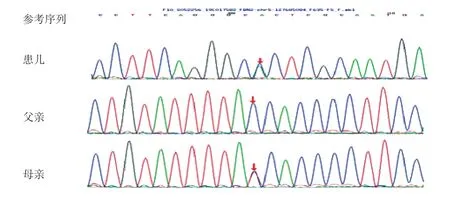
图3 患儿FBN2 基因测序图(c.2944T >G,p.C982G)
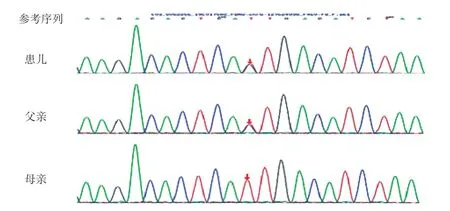
图4 患儿FBN2 基因测序图(c.6518A >G,p.N2173S)
2 讨论
BHS 是一种极其罕见的常染色体显性遗传病,由Epstein 等[4]在1968 年首次报道,继而由Beals 和Hecht 在1971 年与MFS 进行了鉴别[5]。目前文献报道FBN2基因是唯一已知的与BHS相关的致病基因。BHS 临床表型与MFS 类似,但是基因型不同,BHS是由FBN 2基因变异引起,而MFS 是由定位于染色体15 q 15-21 的FBN 1基因变异引起。因BHS 临床表型与MFS 有重叠,BHS 的发病率尚不清楚,患病率亦很难估计,但在鉴定出FBN 2基因变异后,报告的患者人数有所增加。目前人类基因数据库中,在72 例诊断为BHS 的患儿中有63 例存在FBN 2基因变异[6]。
FBN 2基因定位于染色体5 q 23-q 31,由65 个外显子组成,编码纤维蛋白2(2 912个氨基酸)。对于如此大的一个基因库,到目前为止仅证实了为数不多的变异。截止2016年5月20日,Clinvar等只记录了336种变异[7]。根据HGMD 数据库显示有51 种变异具有致病性,其中54%的致病变异为错义变异,表现为半胱氨酸被其他氨基酸替代,结果导致原纤维蛋白2 结构和功能改变。有报道发现FBN2基因新的错义变异c.3229 T>G(p.C1077G)[8]。本例患儿的2个变异位点c.2944T>G(p.C982G)和c.6518A>G(p.N2173S)为新的错义变异。
FBN 1、FBN 2 和二聚体是细胞外基质的主要成分,它们能形成微纤维,从而为弹力组织提供形成弹力支架。而FBN 2 与组织韧性有关,在连接机体关节和器官结缔组织中对于能提供硬度和韧度的弹力纤维细胞外微纤维的形成发挥至关重要的作用[9]。FBN 2 主要在胚胎早期形成的弹性软骨、主动脉中膜和支气管上皮中表达,其发生异常将会影响细胞外基质中微纤维10 nm 的延伸[10]。同时,FBN 2基因变异可减少原纤维蛋白2 的形成或形成功能异常的原纤维蛋白2,而微纤维形成减少将会降低纤维韧性,患者表现为前肢挛缩、双侧并指等临床症状[11]。
BHS和MFS具有许多共同临床特征,例如外貌特征,包括高瘦、外表羸弱和骨骼特征(如蜘蛛样指、细长指、漏斗胸和脊柱后凸)。但BHS的关节屈曲挛缩、皱耳和肌肉发育不良是其特有的,并且MFS通常没有眼部和心血管并发症。国外学者对FBN 1与FBN 2基因变异导致的临床表型进行总结表明,典型MFS、新生儿MFS、BHS和重度BHS患儿可分别表现出不同临床症状,且不同症状间关联性及所占比例有差异(表1)[12]。本例患儿除蜘蛛样指、细长指外,还有四肢屈曲挛缩、皱耳和下肢肌肉发育不良,与文献报道一致。
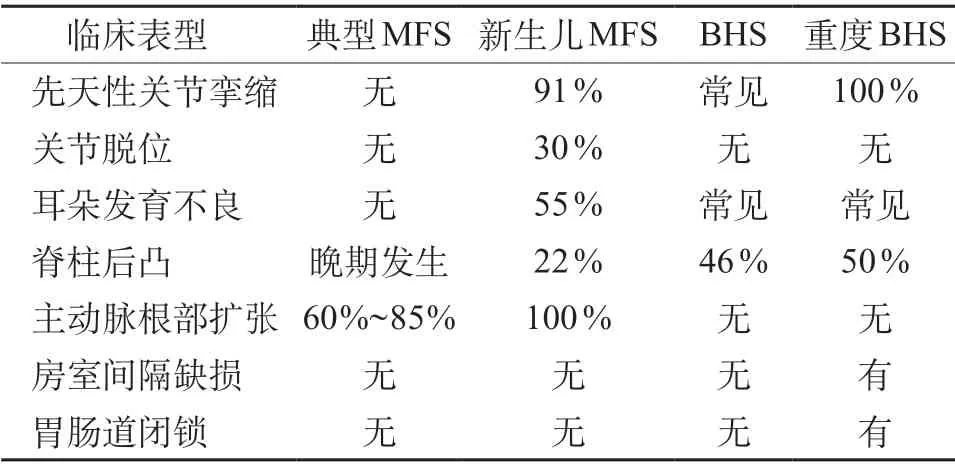
表1 FBN1与FBN2基因变异患儿不同临床表型比较
BHS 患者由于关节挛缩,运动发育迟缓很常见。越来越多报道发现,由FBN 2变异导致的BHS 患者心血管系统受累亦较为常见,如先天性心脏病、主动脉根部扩张、二尖瓣关闭不全、二尖瓣脱垂等[13],但BHS 患者最严重的并发症是脊柱侧弯畸形。据报道20%的BHS患者可出现眼科异常,包括异位症、蓝色巩膜、青光眼,部分出现晶状体缺损、轻度白内障和睫状体异常[14]。
迄今为止,BHS 尚无统一的临床诊断标准,确诊需依靠基因检测。BHS的治疗主要是对症治疗。出生时不同程度的关节挛缩可能随着时间趋于减轻,但弯曲畸形总是存在。脊柱后凸畸形通常在儿童早期出现,常规体格检查可发现脊柱畸形,尽早干预可以预防以后的发病,如严重畸形时须进行脊柱后凸矫正手术。运动发育迟缓需定期进行康复训练及接受个性化教育。因BHS可出现眼部及心血管系统并发症,建议进行全面的眼科评估及连续超声心动图检查评估心脏受累情况。
综上所述,本例患儿经基因检测确诊为BHS。BHS 可表现为多系统、多器官受累,且其临床表型与MFS极其类似,两者鉴别依赖基因检测。
