CRLF1 复合杂合变异致Crisponi 综合征1 例临床特征
2021-09-13刘银芝廖镇宇杨志明叶红球占彩霞黄瑞文
刘银芝 廖镇宇 杨 慧 杨志明 叶红球 占彩霞 黄瑞文
湖南省儿童医院新生儿科(湖南长沙 410007)
Crisponi综合征/冷诱导出汗综合征-1型(Crisponi syndrome/cold induced sweating syndrome type 1,CS/CISS1)是一种常染色体隐性遗传性疾病,由细胞因子受体样因子1(cytokine receptor like factor 1,CRLF1)基因变异引起。1978 年首次报道一对以色列姐妹在凉爽环境中背部和胸部大量出汗,同时合并上颚高拱、鼻梁塌陷、肘部不能充分伸展、脊柱后凸,命名为CISS[1]。1996年发现在意大利撒丁岛地区17例新生儿具有相同的特征,如哭吵或刺激后出现面部肌肉痉挛、喂养和呼吸困难等,命名为CS[2]。2007年经基因测序等发现CS与CISS为同一基因变异所致,CS是CISS1婴儿期的表现[3]。撒丁岛是世界上CS/CISS发病率最高的地区,通过等位基因联合携带频率计算出预计发病率约1/20 700[4]。中国尚未见报道,现报告1 例因CRLF1基因新发复合杂合变异致CS,分析其临床表型和基因型,并复习相关文献。
1 临床资料
男性患儿,出生50 小时,因张口困难、哭声小伴呼吸困难入院。患儿系G2P2,足月剖宫产,出生体质量3.5 kg,无窒息抢救病史,羊水、胎盘、脐带均无异常;Apgar评分9-10-10分。有一哥哥,6岁,身体健康。无家族遗传及特殊病史。患儿于生后半小时即发现有张口困难,无法吸吮母乳,哭声小、呼吸困难、口吐泡沫,无抽搐及发热。入院体格检查:体温无异常,心率140次/min,呼吸39次/min,体质量3.4 kg;反应欠佳,哭声小、不连续,头颅无包块;前囟平软,皮肤轻度黄染,无皮疹;口唇无发绀,口腔不能完全打开,小下颌;呼吸欠规则,双肺呼吸音粗,有痰鸣音;心前区无隆起,心律齐,胸骨左缘2~3肋间2/6级收缩期杂音;腹部无异常;手、足趾指细长,且肘关节、双手中指、无名指屈曲,不能伸直(图1);四肢肌张力增高;拥抱反射、握持反射引出不完全,吸吮反射、觅食反射未引出。实验室检查:血常规白细胞9.81×109/L;中性粒细胞37.1%;淋巴细胞43.8%,血红蛋白132 g/L;血小板75×109/L;C反应蛋白21.16 mg/L,降钙素原0.50 ng/mL;血生化、肝功能、电解质、血脂、血气分析、血糖均未见明显异常;TORCH IgM抗体均阴性。脑脊液常规、生化、培养未见异常。四肢长骨X线摄片:骨质未见明显改变。心脏彩超示卵圆孔未闭。胸片示新生儿肺炎改变。彩超检查:脑实质回声增强(PVE Ⅱ度),双侧侧脑室后角增宽,双肾实质回声增强,双侧睾丸鞘膜腔积液。患儿入院后予吸氧及抗感染治疗10天,因患儿吞咽功能差,无吸吮反射,喂养困难,最终家长放弃治疗,出院后死亡。

图1 患儿手部特征
为进一步明确病因,经医学伦理审核,患儿父母知情同意,住院期间采集患儿及父母外周血,送至长沙金域医学检验实验室利用Illumina HiSeq2000平台进行全基因组编码外显子高通量测序,并经Sanger测序验证。结果显示,患儿CRLF1基因第5号外显子区存在2个变异:c.829_847del p.(Arg277Thrfs),遗传自父亲;c.713delC p.(Pro238Argfs),遗传自母亲(图2)。2个变异均未见文献报道,HGMD数据库和clinvar数据库均未收录,千人基因组(1000g2015aug_ALL)、ESP6500siv2_ALL、ExAC_ALL、gnomAD_genome_ALL 和dbSNP 147 数据库均未见收录。根据《美国医学遗传学与基因组学学会新版指南》,c.829_847 del(p.Arg277Thrfs)为移码变异,预计会使所编码的蛋白质自第277 位氨基酸Arg 开始发生编码紊乱,导致所编码的蛋白质发生截短从而丧失其正常功能(PVS1),同时正常对照人群数据库(Exome Sequencing Project EAST,1000 Genomes Project等数据库)中均未发现该变异(PM 2);与检测到的另一变异分别位于不同的等位基因(PM 3),判定其为致病变异。c.713 delC(p.Pro238Argfs)也为移码变异,预计会使所编码的蛋白质自第238位氨基酸Pro开始发生编码紊乱,导致所编码的蛋白质发生截短从而丧失其正常功能(PVS1),同时正常对照人群数据库(Exome Sequencing Project EAST,1000 Genomes Project等数据库)中均未发现该变异(PM 2),判定其为可能致病变异。上述2 个致病变异为复合杂合变异,符合常染色体隐性遗传规律,患儿可以确诊为CRLF1基因变异致的CS。
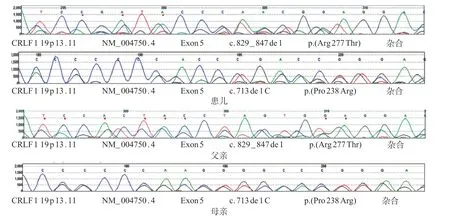
图2 患儿及父母CRLF1 基因测序图
2 讨论
CISS1和CS是由同一种基因CRLF1变异导致的常染色体隐性遗传性疾病,CS是CISS1婴儿期的表现。CS是导致新生儿猝死的重要原因之一,典型临床特征有:喂养和吞咽困难、呼吸困难、发热、面部肌肉痉挛、屈曲指及特殊面容,特殊面容包括圆脸、丰满的脸颊、突出的颧骨、宽鼻、前鼻孔、长剑突、低耳位、小嘴、小下颌、短颈、低发际线。大多数CS患儿在生后第一年死亡,存活个体为CISS,表现出发热和面部肌肉收缩自发缓解,约3岁左右,在环境温度低于20 ℃时,上身、面部和手臂大量出汗,这与血浆肾上腺素水平大量增加有关,同时伴有脊柱侧弯和轻度的精神运动发育迟缓。
CRLF1作为CS致病基因首次在2003年报道,基因定位于19 p 13.11 号染色体,由9 个编码外显子组成,跨度为14 kb,并被转录为1 824 bp的线性mRNA,编码一个由422个氨基酸组成的蛋白质(46 kDa)[5]。CRLF1/CLCF1复合物结合到CNTFRα启动GP130、LIFR二聚化,刺激JAK/STAT信号传导通路。CNTFR通路对面部运动神经元的胚胎发育和支配汗腺的交感神经元的胆碱能分化具有重要意义。CLCF 1/CRLF 1 介导支配汗腺的交感神经元的肾上腺能向胆碱能的转变,且胆碱能交感神经还支配骨骼肌的骨膜和血管系统。肾上腺素能转化为胆碱能表型的缺陷可能解释骨畸形和肌肉症状,以及不同生命阶段CS/CISS 临床表现的高变异性。CRLF 1基因可能还存在其他功能,需进一步研究[6-7]。目前发现的CRLF 1基因变异已有40余种,虽然大多数变异热点是发生在单独个体,但也有一定地域性,如 c.226T>G和c.676_677 dupA 主要发生在撒丁岛,c.708_709 delinsT 主要发生在土耳其,c.983 dupG 主要发生在沙特阿拉伯和c.713dupC主要发生在西班牙[4]。
本例患儿出生后即起病,有部分典型临床特征,如手指屈曲畸形、呼吸困难、喂养困难、小口及小下颌,同时存在四肢肌张力增高、张口困难。高通量测序行全外显子分析,最终发现患儿为CRLF1基因新复合杂合变异导致的CS。
迄今为止,中文文献尚无CS/CISS1报道,国外报道98例,其中83例为CRLF1基因变异,另15例撒丁岛患者未行基因分析。大部分CS/CISS1来自欧洲,尤其是地中海区域(撒丁岛27例、土耳其23 例、沙特阿拉伯13例、西班牙8例、意大利4例)[3-5,8-29]。主要临床表型依发生率为:吞咽困难、发热、面部肌肉收缩、屈曲指畸形、唾液分泌过多、胖胖的圆脸颊、下颌活动受限、发绀、牙关紧闭、低鼻梁、高腭弓、颈部肌肉紧张。虽然CS/CISS1在全球的发生率很低,但在土耳其等地发生率较高。目前CS/CISS1治疗无特异性方法,主要为对症支持治疗、矫正畸形,通过个体化管理,以延长生存时间,促进生长发育,尽量减少疾病对家庭生活的影响。新生儿期的治疗非常关键。CS新生儿需要一个相对封闭的环境,比如给予患儿一个黑暗的安静的房间,减少操作,必要的操作尽可能温和集中进行,尽量减少护理刺激。室温或暖箱的温度不应超过20 ℃。体温调节功能障碍导致新生儿对退热药没有反应,只能通过物理降温。新生儿需要持续监测心率、呼吸、血氧饱和度和体温,以避免出现呼吸困难、高热等危及生命。新生儿出院后可使用脉搏血氧饱和仪进行持续监测,由于喂养问题经常需要使用鼻饲管或经皮内镜下胃造瘘术。CISS1个体还需要处理进行性脊柱侧弯、畸形以及随环境变化的出汗。可乐定属于咪唑啉类活性物质,是一种α2-肾上腺素受体激动剂,通过G蛋白介导的信号通路通过反馈抑制减少突触去甲肾上腺素的释放,从而导致交感神经活性降低而减少出汗,可减少冷诱导出汗。与可乐定类似,莫索尼定是咪唑啉1 型受体激动剂,通过减少突触去甲肾上腺素的释放,引起交感神经活性的减低。有报道莫索尼定有效缓解2例CISS1的临床症状[10]。
本例患儿新生儿期起病,有部分典型临床特征,实验室检查结果无特殊表现,但无典型的发热表现,与国外报道病例特点不完全一致,可能受地域、遗传等各方面因素的影响。本例患儿CRLF 1基因存在新发变异c.829_847 delp.(Arg277 Thrfs)和c.713 delC p.(Pro238 Argfs),扩展了CRLF1致病变异谱,国内未见报道。
