10q22.3q23.3 微缺失综合征合并高甲硫氨酸血症1 例报告并文献复习
2021-09-13王彦云
王彦云 孙 云 蒋 涛
南京医科大学附属妇产医院(南京市妇幼保健院)遗传医学中心(江苏南京 210004)
10q 22.3q 23.2 微缺失综合征(10q 22.3q 23.2 microdeletion syndrome)是一种罕见的单基因遗传病,呈常染色体显性遗传,目前暂无流行病学数据。美国于1999年首次报道并描述[1],截止当前,PubMed外文数据库中已有16 例报道[1-8]。已报道患者呈散发性,多为自发变异。临床特点是智力发育落后、面部畸形、肠息肉等。
高甲硫氨酸血症(hypermethioninemia)已被纳入我国新生儿串联质谱筛查病种,高甲硫氨酸血症对应的致病基因MAT1A位于10q 22.3(80270002-80291821)。当10q 22.3q 23.2 微缺失综合征累及MAT1A基因时,如MAT1A对侧等位基因合并致病性变异,可造成10q22.3q23.2微缺失综合征患儿合并高甲硫氨酸血症。本文报道1 例10q 22.3q 23.2 微缺失综合征合并高甲硫氨酸血症病例,类似病例尚未见报道。
1 临床资料
回顾分析1例在2018年因新生儿串联质谱筛查蛋氨酸(methionine,MET)升高,于南京医科大学附属妇科医院最终确诊为10q22.3q23.2微缺失综合征合并高甲硫氨酸血症患儿的临床资料。
患儿为男性,G2P2,足月剖宫产,出生体质量4.3 kg。出生后72 小时,采集患儿足跟血制成干血斑滤纸片样本,应用串联质谱(tandem mass spectrometry,MS/MS)检测系统(Waters TQD)进行氨基酸谱和酰基肉碱检测分析行新生儿筛查。结果提示MET 306.02 μmol/L,召回于出生第10天复查MET升至695.37 μmol/L。患儿有特殊面容,额突出、鼻根低、眼距宽、内眦赘皮、鼻头轻度上翘、人中浅、耳位低,余未见特殊异常。肝肾功能、血清同型半胱氨酸、电解质、血尿便常规均无异常。头颅磁共振成像(MRI)示双侧大脑半球对称,灰白质对比可,各脑室、脑池大小形态正常,双侧小脑半球脑白质对称斑片样T2 WI高信号(图1),脑干未见明显异常,DWI 未见弥散受限。听力初筛通过。
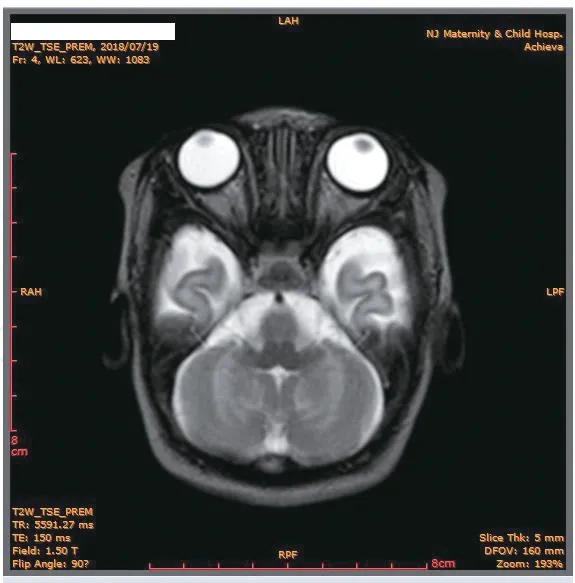
图1 患儿颅脑MRI 表现
为明确患儿MET升高的原因,经医学伦理审核以及患儿父母知情同意,采集患儿及亲属外周血各2 mL提取DNA,行高通量二代测序,并重点对MAT1A、GNMT、ADK、AHCY、CBS、MTR和MTHFR共计7个基因的序列进行生物信息学分析,其中MAT1A基因9 个外显子(编码区长度1 188 bp)覆盖度100%,具体检测方法参考文献[9]。检测结果示,患儿MAT1A基因c.74_75 delTG(p.Val 25 GlyfsX 7)纯合变异(表1)。

表1 先证者高通量测序检测到的变异详细结果
随后针对MAT1A基因进行荧光定量PCR 检测,MAT1A基因共9个外显子,选取第1、5、9号外显子进行检测,具体方法参考文献[10]。结果显示,患儿及其父亲MAT1A基因第1、第5 和第9 号外显子均存在杂合缺失,即这3个外显子均为单拷贝。
进一步采用拷贝数变异(copy number variation,CNV)分析明确缺失位置及片段大小。使用商品化CNV检测文库构建试剂盒,通过Illumina测序平台检测CNV,具体检测方法参考文献[10]。患儿及其父亲CNV检测示arr[hg19]10q22.3q23.2(81,622,294-89,253,430)×1,缺失片段大小为7.63 Mb。患儿父亲为单纯10q22.3q23.2微缺失综合征,其缺失片段为自发变异,患儿缺失片段遗传自其父亲。
患儿姐姐临床表型无异常,未进行基因检测,其他亲属,包括爷爷、奶奶、父亲和母亲的基因组验证分析确认患儿父亲为10q 22.3q 23.2 微缺失综合征,其缺失性CNV为自发变异(de novo),患儿母亲携带c.74_75 delTG(p.Val 25 GlyfsX 7)杂合变异(图2)。患儿于2 月龄时最终确诊基因型为10q 22.3q 23.2 deletion/c.74_75delTG(p.Val25GlyfsX7)。
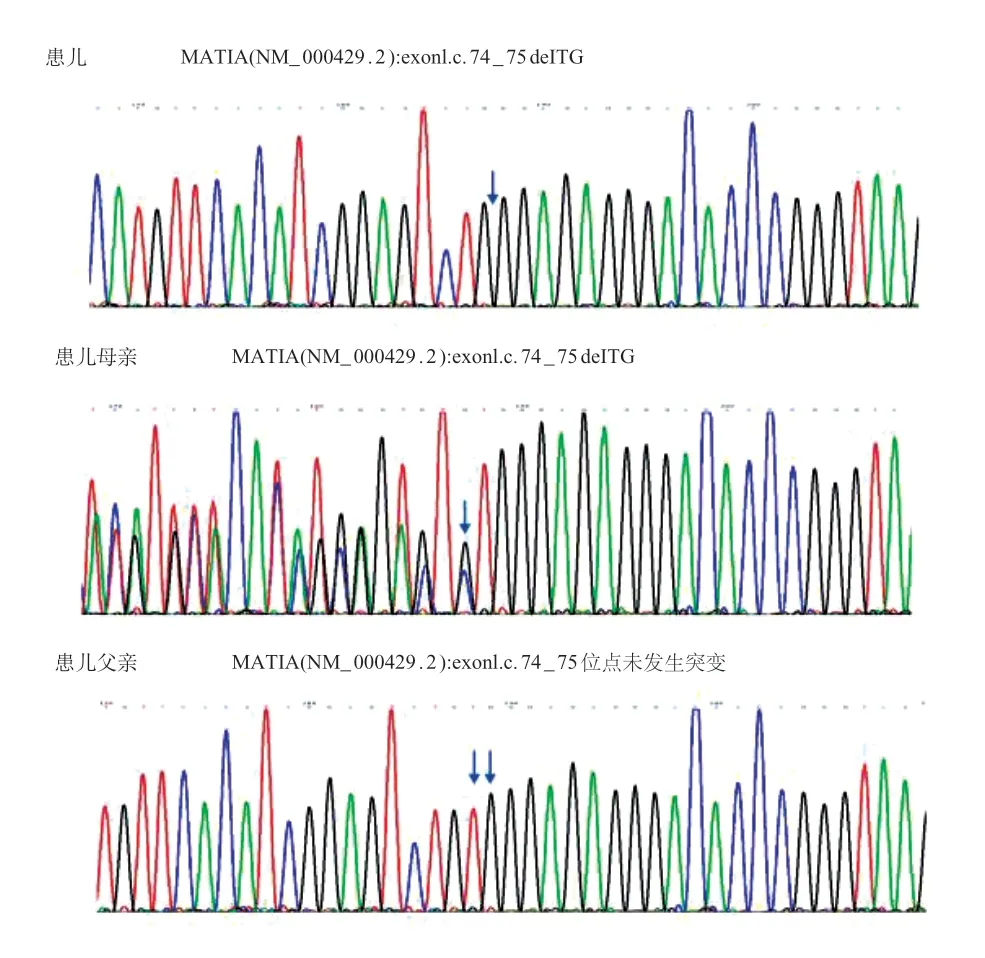
图2 患儿及其父母家系Sanger 测序验证结果
限制患儿蛋白质饮食,婴儿期予不含MET 的氨基酸配方结合母乳喂养,添加辅食后低蛋白饮食,坚持康复训练,定期串联质谱检测复查MET。患儿末次随访为2岁5个月。患儿运动发育基本正常,7月龄可独坐,15月龄可独走,当前可听懂并执行简单的指令,但语言发育迟缓,仅能发“爸”“妈”等单音节字。患儿8月龄前,MET控制在200 μmol/L左右;9月龄后,饮食控制依从性差,MET仅能控制在700 μmol/L左右,肝功能基本正常。头颅MRI未复查。
患儿父亲身高161 cm,特殊面容,额突出,鼻根低,眼距宽,内眦赘皮,鼻头轻度上翘,人中浅,耳位低,双手杵状指,余未见特殊;自幼语言及运动发育落后,易患上呼吸道感染。当前患儿父亲存在性功能障碍(患儿姐姐及患儿均为借助辅助生殖技术妊娠),沟通理解力稍差,存在胃肠道易激惹征(每餐进食后30分钟均立即排便,大便不成形)。结肠镜检无异常;血常规、肝肾功能、心电图、腹部彩超等均未见明显异常;串联质谱检测MET 26.6 μmol/L。
2 讨论
以“10q 22 .3q 23 .3 微缺失综合征”“高甲硫氨酸血症”“10q 22.3q 23.3 microdeletion syndrome”“10q 22.3q 23.3 deletion syndrome”“Hypermethioninemia”为关键词,检索万方数据知识服务平台、中国知网、PubMed 文献数据库建库至2020 年5月。结果显示,从外文数据库中共检索到16例患者,均有明显的颅面畸形特征;其他临床表现差异较大,智力发育从基本正常到明显发育迟缓,提示10q 22.3q 23.2 片段缺失外显不完全,但目前报道的临床表现中暂无性功能和杵状指的描述。鉴于10q22.3q23.2缺失综合征外显不全,本例患儿的预后不能完全等同于其父亲,需要更长的时间进行随访。此外,10q22.3q23.2 片段中包括BMPR1A基因,而据文献报道BMPR 1 A基因杂合变异可引起幼年性息肉病综合征。患儿父亲合并有胃肠道症状,但肠镜检查未发现息肉。本例患儿后续随访中也应加强肠道症状观察及相应检查。
10q22.3q23.2微缺失综合征自1999年首次被作为一种疾病报道后,目前外文数据库中已报道16 例。由于存在外显不全,16 例患者除均有特殊面容外,临床表现差异较大。本例患儿的父亲为自发变异、单纯10q22.3q23.2微缺失综合征,其特殊面容、幼年期易罹患上呼吸道感染、运动和智力发育迟缓、胃肠道症状均与文献报道相吻合,但是患儿父亲的杵状指及性功能障碍均为首次描述。本例患儿同时也是家系中的10q 22.3q 23.2 微缺失综合征患者,其基因缺陷遗传自父亲,虽然患儿具有与其父亲类似的特殊面容,但大运动发育正常,大便正常,无胃肠道易激惹征象,与其父亲临床表现差异大,符合外显不全的特点。患儿成年后是否会出现杵状指、性功能障碍等症状还需长时间的随访观察。
近年来,串联质谱(MS/MS)技术在遗传代谢病筛查领域的引入,极大扩展了新生儿筛查项目的疾病种类,其中包括高甲硫氨酸血症。高甲硫氨酸血症的致病基因为MAT1A基因,位于10q 22.3,与10q22.3q23.2微缺失综合征的基因缺陷范围有重合。因此理论上,10q 22.3q 23.2 微缺失综合征患者可能合并有高甲硫氨酸血症。本例患儿是10q22.3q23.2微缺失合并高甲硫氨酸血症,同时罹患此2 种遗传代谢病的临床病例既往未见报道。
在早期的研究中,高甲硫氨酸血症被认为是一种良性疾病。然而,最近的研究表明,过高的MET 浓度会导致肝脏和中枢神经系统损伤[11-13],建议高甲硫氨酸血症患者在MET 浓度高于800 μmol/L 时,应采用低MET 饮食治疗,使MET 水平保持在500~600 μmol/L 之间。本例患儿在8 月龄前通过低MET饮食治疗,将MET控制在200 μmol/L左右,但是9月龄后,随着辅食摄入增加,低MET联合低蛋白饮食仅能将MET控制在700 μmol/L。目前临床缺乏能有效降低MET的药物,仅以饮食治疗为主,疾病进展后期可行肝移植手术。
此外,本例患儿高甲硫氨酸血症的诊断依赖高通量测序技术的发展,当前,高通量测序技术已被常规应用于遗传性代谢病的基因诊断。但是,本例患儿的第一次高通量测序结果错误,测序结果显示MAT1A基因c.74_75 delTG纯合子变异。进一步的遗传分析表明,MAT1A基因在其他等位基因上存在缺失。这是由于基于高通量测序的Panel不能灵敏地识别大片段结构变异,如大片段缺失。结合基于高通量测序的Panel和PCR的技术或CNV技术有助于获得正确的基因诊断。
