两种高通量测序平台应用于不同SARS-CoV-2变异株的对比研究
2022-10-19李东晓马红霞王海峰黄学勇郭万申
李东晓,李 懿,朱 琳,宋 云,马红霞,王海峰,叶 莹,黄学勇,郭万申
高通量测序,又名下一代测序(Next Generation Sequencing,NGS),可直接对人体临床样本中的核酸进行测序,实现对感染性疾病的检测、分型及溯源,为促进分子流行病学研究和公共卫生事件调查等多个方面提供助力[1]。新型冠状病毒肺炎(Corona Virus Disease 2019,COVID-19)首次被发现就是基于二代测序的mNGS技术,截止2022年2月,全球报告COVID-19确诊病例超过4亿[2-3]。为了实时监测新型冠状病毒的快速变异及演化规律,高通量测序技术在新型冠状病毒的鉴定、分型、溯源方面发挥着关键作用,短读长的二代测序一直是病原基因组学的金标准,三代测序作为新型快速诊断技术,在实验周期、成本及便携性等方面扮演补充角色[4-5]。本文利用Illumina MiSeq和Oxford Nanopore两种测序平台分别对2021年境外输入的4例COVID-19病例以及2022年本地6例COVID-19病例的上呼吸道样本进行全基因组测序,对比分析这两种测序平台在新型冠状病毒溯源分析研究中的特点。
1 材料与方法
1.1 样本来源 10份上呼吸道样本来自确诊COVID-19的病例,采集时间为2021年6月至2022年1月,其中包括4例境外输入病例、6例本地病例,样本于-80 ℃保存。
1.2 仪器与试剂 新型冠状病毒核酸检测试剂盒(伯杰,中国上海);实时荧光定量PCR仪(LightCycler 96,瑞士罗氏公司);PCR扩增仪(BIO-RAD T100,美国伯乐公司);Qubit荧光定量仪(Qubit 3.0,美国赛默飞公司);高通量测序仪(Illumina MiSeq,美国Illumina公司);测序芯片(FLO-MIN106D,英国牛津纳米孔公司);GridION Mk1测序仪(Oxford Nanopore,英国牛津纳米孔公司)。
1.3 建库前处理 按核酸提取试剂盒说明书(天隆,中国西安)步骤进行核酸提取操作,得到10份样本的总RNA。样本S9和S10提取后的核酸用无核酸酶水进行梯度震荡混匀稀释并编号,样本S9核酸稀释为S11、S12、S13;样本S10核酸稀释为S14、S15、S16,所有核酸均用伯杰的新型冠状病毒核酸检测试剂进行荧光PCR检测,样本S9和S10的稀释倍数及样本编号见图1。

图1 样本S9和S10核酸稀释处理流程Fig.1 Nucleic acid dilution processing steps (sample S9 and S10)
1.4 二代测序全基因组文库构建和测序 采用北京微未来科技有限公司的新型冠状病毒全基因组捕获试剂盒(V-090418-1)将提取的病毒总RNA进行逆转录和特异性扩增。利用Invitrogen公司核酸定量分析试剂盒(Q32854,美国)对扩增后的cDNA进行定量,使用美国Illumina公司的Nextera XT文库制备试剂盒(FC-131-1024)构建测序文库并进行磁珠纯化(A63880,Beckman Coulter公司,美国),最后用Illumina公司的基因测序试剂盒(MS-102-2002,美国)在MiSeq测序仪进行全基因组测序。
1.5 三代测序全基因组文库构建和测序 取样本扩增纯化后的cDNA进行核酸定量,取100 ng为模板,样本S1、S7~S10依次按说明书使用英国牛津纳米孔公司的连接测序试剂盒(SQK-LSK109)、无扩增条码试剂盒(EXP-NBD114)进行建库;样本S2~S6使用牛津纳米孔公司的快速建库条码测序试剂盒(SQK-RBK004,英国)制备文库,利用牛津纳米孔公司的测序引发试剂盒(EXP-FLP002,英国)对测序芯片预处理,加入待测文库后,运行GridION基因测序仪进行全基因组测序。
1.6 数据分析 以NCBI(National Center for Biotechnology Information)中SARS-CoV-2全基因组序列(Wuhan-Hu-1株,GenBank:MN908947)作为参考序列,使用德国凯杰公司的CLC Genomics Workbench 21.0软件对测序原始下机数据进行序列拼接,得到的序列在Nextclade(https://clades.nextstrain.org/)和Pangolin(https://clades.nextstrain.org/)在线分析工具进行分型,运用MEGA-X软件进行变异位点分析。
1.7 统计学分析 采用美国IBM公司的SPSS 22.0软件进行统计学分析,满足正态性分布采用(均数±标准差)进行统计描述,不服从正态分布采用中位数(四分位数间距)进行统计描述。对10份样本的二代测序和三代测序覆盖度比较采用Wilcoxon秩和检验;8份样本三代测序不同时间覆盖度采用单因素方差分析的Welch分析。P<0.05为差异有统计学意义。
2 结 果
2.1 全基因组测序结果 10份样本进行荧光定量PCR检测,ORF1ab基因Ct值分布在13.2~30.98之间。10份样本二代和三代测序分型结果一致,按照Pangolin分型法分为3个型别,S2、S3、S4为Omicron(BA.1)变异株,二代测序基因组序列全长29 873 bp,三代测序基因组全长29 873~29 882 bp;S9为Alpha(B.1.1.7)变异株,二代测序基因组全长29 869 bp,三代测序基因组全长29 868 bp;其余6份样本均为Delta(B.1.617.2)变异株,二代测序基因组序列全长29 862~29 891 bp,三代测序基因组全长29 867~29 891 bp。二代和三代测序均分型成功,分型结果见表1。
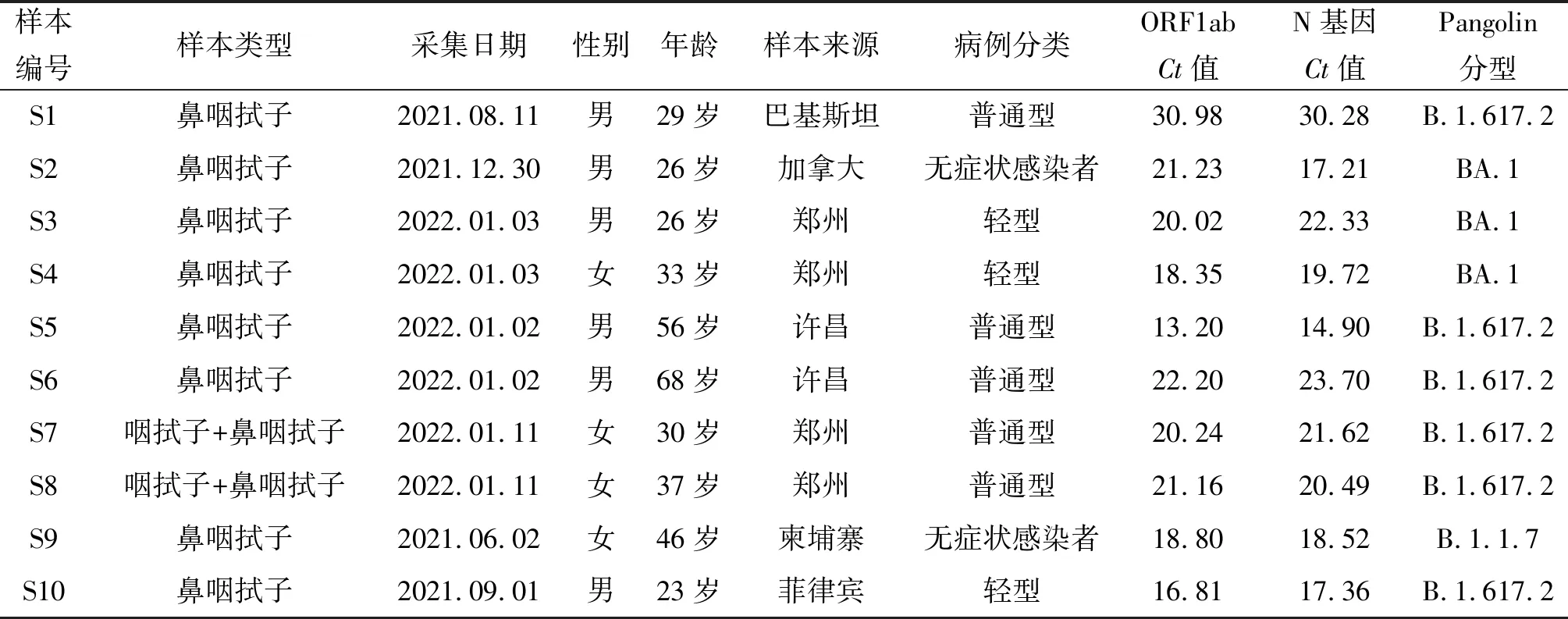
表1 10份样本的全基因组序列分型Tab.1 Whole genome sequence typing of ten samples
2.2 二代和三代测序序列对比 Illumina和Nanopore两个测序平台的覆盖度显示,样本S7的二代测序覆盖度最低(98.48%),其余样本在两个平台的覆盖度均能达到99%以上。二代测序覆盖度中位数为99.73%(0.36%),三代测序覆盖度中位数为99.90%(0.37%),10份样本的二代和三代测序覆盖度差异有统计学意义(t=-2.037,P<0.05)。Illumina平台测序时长达24 h左右,平均测序深度(11 866.8±5 781.9);Nanopore平台测序时间为6~21 h不等,平均测序深度为1 257.5(3 137)。二代测序样本S4(Ct值:18.35)平均测序深度最高(19 209),样本S1(Ct值:30.98)测序深度最低(4 151);三代测序样本S10(Ct值:16.81)平均测序深度最高(4 410),样本S6(Ct值:22.2)平均测序深度最低(533)。两种测序平台比较见表2。
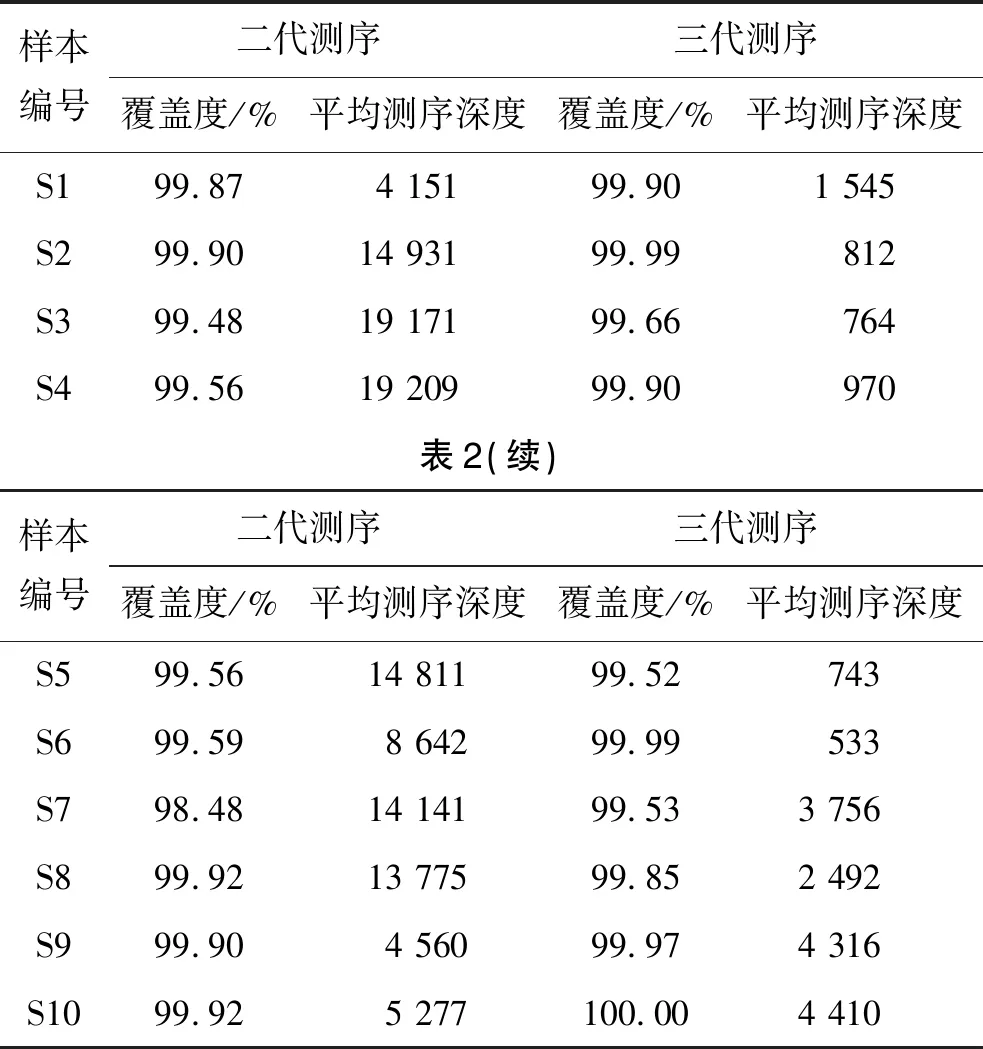
表2 两种测序平台覆盖度及平均测序深度比较Tab.2 Comparison of coverage and mean sequencing depth between sequencing platforms
2.3 不同变异株的变异位点分析 与参考基因组相比,Alpha变异株二代测序检测到41个核苷酸变异位点;4份Delta变异株检测到47个核苷酸变异位点,样本S1检测到42个,样本S10检测到35个;Omicron变异株分别检测到55个、61个核苷酸变异位点。Alpha变异株S基因编码区涉及到非同义突变有8个,Delta变异株涉及到的非同义突变9~11个,Omicron突变株涉及到的非同义突变30个。所有样本均出现D614G的变异,位点变异情况详见表3。
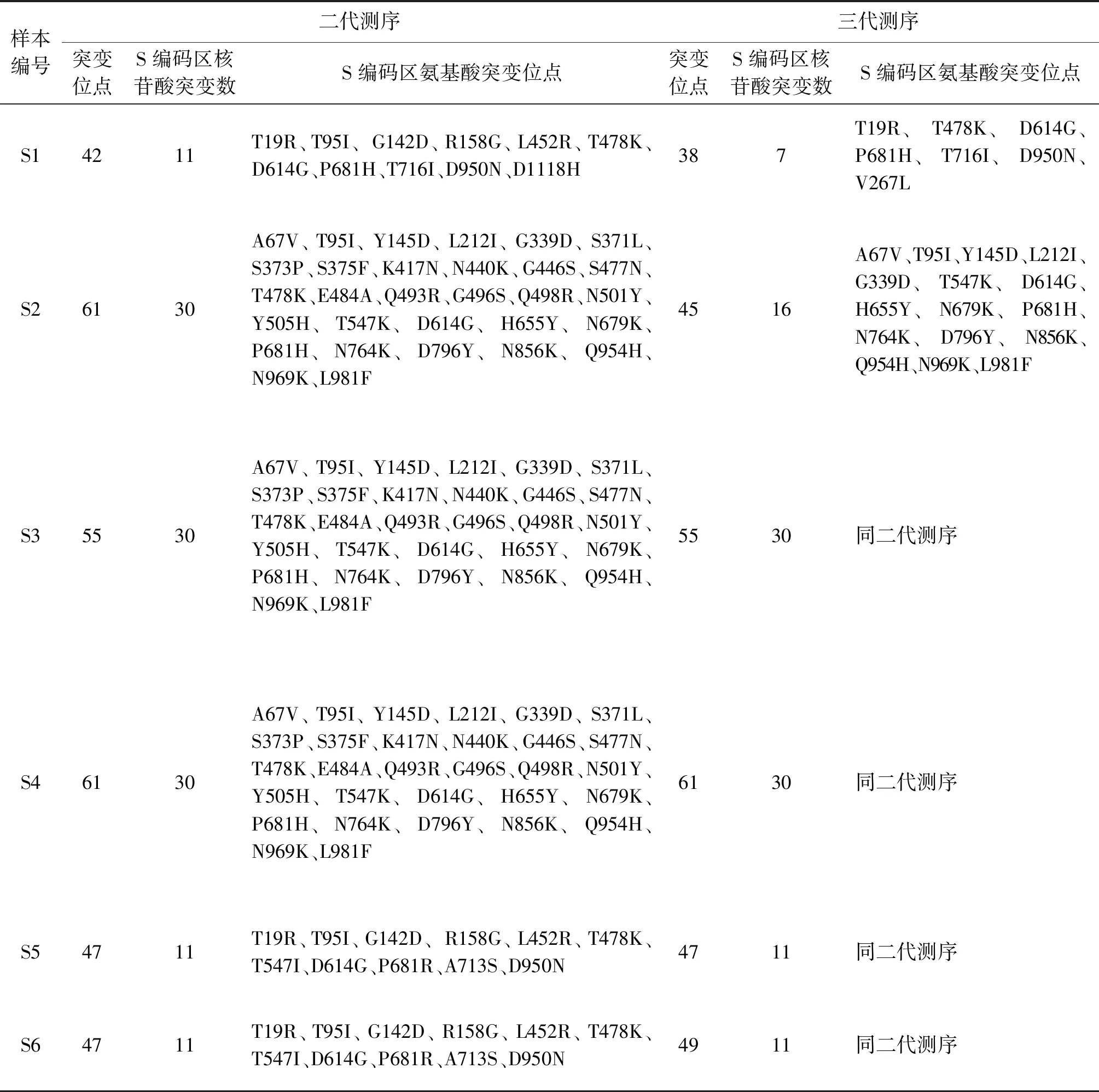
表3 两种测序平台位点变异比较Tab.3 Comparison of nucleic acid and amino acid sequences between sequencing platforms

表3(续)样本编号二代测序三代测序突变位点S编码区核苷酸突变数S编码区氨基酸突变位点突变位点S编码区核苷酸突变数S编码区氨基酸突变位点S74710T19R、T95I、G142D、R158G、L452R、T478K、T547I、D614G、P681R、D950N 4710同二代测序S84710T19R、T95I、G142D、R158G、L452R、T478K、T547I、D614G、P681R、D950N 4710同二代测序S9418V70I、N501Y、A570D、D614G、P681H、T716I、S982A、D1118H418同二代测序S10359T19R、G142D、R158G、A222V、L452R、T478K、D614G、P681R、D950N388T19R、G142D、A222V、L452R、T478K、D614G、P681R、D950N
2.4 不同稀释度样本二代和三代测序对比 Illumina平台所有样本的测序时间均相同,不同稀释度的样本平均测序深度和覆盖度如表4所示,样本S9和样本S10为原样,其余6份稀释样本的Ct值分布在22.69~35.37之间。测序覆盖度最高的是样本S9和S10(Ct值<20);平均测序深度最高的是样本S11(Ct值:24.65)和样本S14(Ct值:22.69);平均测序深度和覆盖度均较低的样本S13和S16,Ct值33~35之间。样本S9、S11~S13的平均测序深度为11 359.5±7 664,样本S10、S14~S16的平均测序深度为11 435.5±6 410.4;不同稀释度样本二代测序覆盖度差异无统计学意义(F=0.091,P>0.05)。
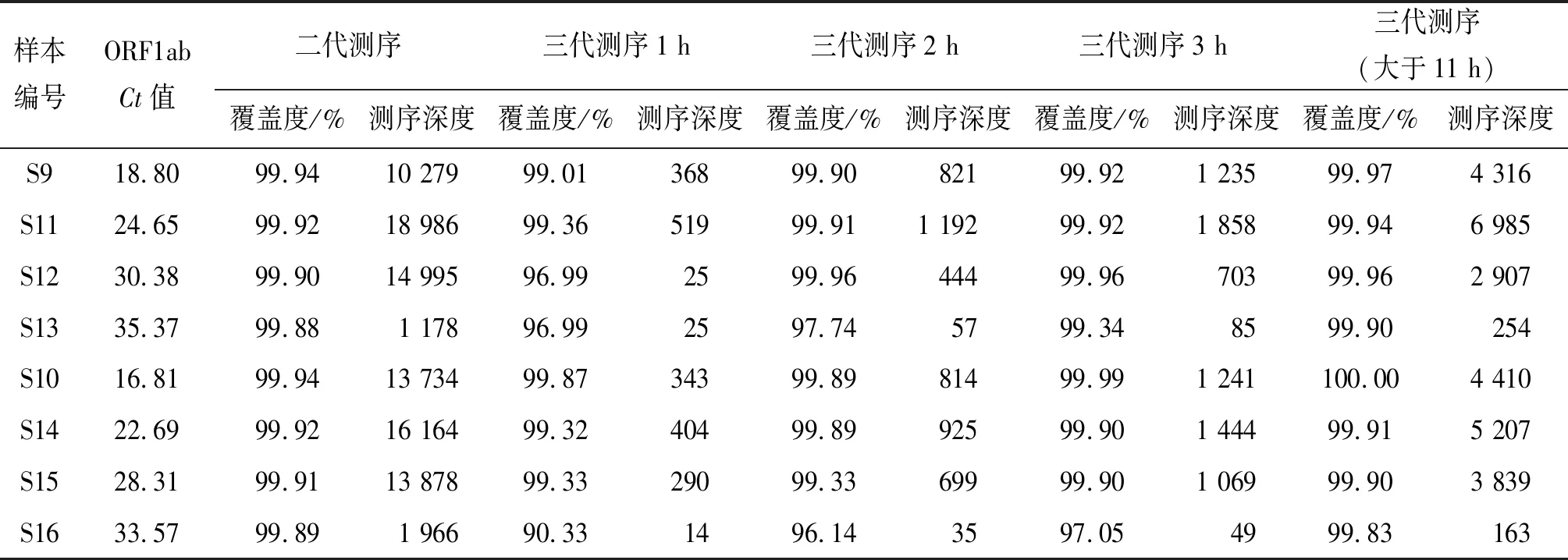
表4 测序不同时间对比Tab.4 Sequencing comparison at various time points
Nanopore三代测序过程中,分别在测序开始后的1、2、3、11 h拷贝数据并拼接分析,在Pangolin在线工具上进行分型,样本S9、S11、S12、S13为B.1.1.7变异株,样本S10、S14、S15、S16为B.1.617.2变异株。所有样本测序2、3、11 h后分型,均与测序1 h的分型结果相同。
测序1 h后,Ct值大于30的样本S12、S13和S16的覆盖度和平均测序深度与其他样本相比均较低;测序2 h和3 h后,样本S12的覆盖度和平均测序深度有所提升;三代测序超过11 h后,样本S13和S16覆盖度分别达到99.90%和99.83%,平均测序深度分别达到254和163,在所有样本中测序深度最低,其他6个样本平均测序深度均大于2 000。三代测序4个不同时间的覆盖度中位数为99.17%(2.36%)、99.89%(1.77%)、99.91%(0.47%)、99.93%(0.07%),差异无统计学意义(F=2.498,P>0.05)。
3 讨 论
目前,WHO一共公布了5种需关注的变异株(VOC),它们分别是:Alpha(B.1.1.7)、Beta(B.1.351)、Gamma(P.1)、Delta(B.1.617.2)和Omicron(B.1.1.529)。Omicron变异株较其它变异株突变位点更多,有更强的传染性和免疫逃逸能力[6],截至2022年2月,Omicron变异株已经至少在142个国家或地区流行,逐渐取代Delta株成为优势毒株[7]。随着新型冠状病毒在全球的不断传播,新冠肺炎病例不断攀升,新的变种不断出现,研究表明,新型冠状病毒基因组每月积累两个单碱基突变[8-9]。基因组监测的大规模应用将有利于更早预测并启动公共卫生策略,遏制SARS-CoV-2变异株及其他新型病毒的暴发[10-11]。
本研究发现,针对同一样本,Illumina二代测序和Nanopore三代测序在覆盖度方面差异无统计学意义,均能获得3种新冠病毒变异毒株的基因序列并准确分型。6份样本S3、S4、S5、S7、S8和S9变异位点保持一致,样本S1、S2的三代测序变异位点总数少于二代测序,样本S6和S10三代测序变异位点数目多于二代测序。样本S1的Ct值为30.98,推测原始样本中病毒部分基因片段数目较少,三代测序测序深度不足导致检测到的变异位点减少。样本S2为Omicron变异株,变异位点较多,S基因编码区扩增效率不高,加之三代平均测序深度(812)远低于二代测序(14 931),变异位点检测总数比二代测序少。样本S6的三代测序比二代测序突变位点总数多2个,分别是ORF1ab编码区T15510C(339C:10T)、N基因编码区G28796A(655A),未引起氨基酸改变。样本S10的三代测序比二代测序突变位点总数多3个,ORF1ab编码区T6552G,对应氨基酸替换M2096R;编码区A18675G、C18676T、T18678C、T18690G为插入缺失,造成2个氨基酸替换R6138C、F6142L。Nanopore单分子测序技术的错误率主要集中在插入缺失,不过这些错误是随机出现的,足够高的覆盖率能在一定程度上弥补该错误率[12]。
S基因编码区突变位点对比发现,样本S3~S9共7份样本的变异位点数目在两个平台保持一致,3份样本(S1、S2、S10)变异位点数目不同,三代测序变异位点检测数目少,二代测序对变异位点的识别更多更精确。针对S基因编码区变异位点不同的样本分析,样本S1缺少5个氨基酸变异位点,S基因编码区增加1个氨基酸变异位点(V267L),对应的核苷酸变异位点G22361T,该位置为杂合位点(2G:3T),因测序深度太低未纳入分析;样本S2和S10三代测序检测的变异位点数均少于二代测序,对应的氨基酸突变数目也相应减少。
为评价Ct值对于测序效果的影响,同时对比不同变异株之间测序有无差别,我们选择两种变异株样本S9(Alpha变异株)和样本S10(Delta变异株),对核酸进行梯度稀释后测序。这两份样本原始Ct值在20以下,通过不断稀释,样本中的病毒载量不断减少,测序结果显示,这8份样本在Illumina二代测序和Nanopore三代测序平台的覆盖度差异无统计学意义;同一样本不同稀释度在两个平台的分型结果保持一致;Ct值33~35的样本,二代测序和三代测序平均测序深度均较低。Løvestad AH等采用Nanopore平台对新型冠状病毒三代测序的研究数据发现,Ct值<33的样本能够保证一致的扩增效率和较高的基因组覆盖度[13]。本次研究16份核酸样本Ct值在13.20~35.37,Ct值<33的样本在二代和三代测序平台均能获得较好的测序结果,证实了以上结论。Lu等[14]的研究也发现,对于Ct值30以上的病毒载量较低的样本,三代测序覆盖度优于二代测序。与短读长的二代测序相比,三代测序的长读长可能更有利于基因组组装和结构变异检测[15]。
Nanopore三代测序采用实时测序的单分子测序技术,实时产出、实时分析是区别于二代的最大特点,对于突发传染病应急检测具有非常重要的作用。对于Ct值<33的样本,三代测序1 h后,平均测序深度达到300以上即可在两个分型平台完成分型,且与之后的分型结果保持一致;Ct值>33的样本,平均测序深度200左右也可以准确分型。从测序成本角度考虑,Ct值较大的样本,随着测序时间延长,新冠病毒基因组大部分序列已被覆盖,一味追求测序深度意义不大,建议测序数据能够拼接分型后停止测序。根据测序芯片不同,Illumina二代测序时间在18~24 h,Nanopore三代测序将大大提升应急检测速度。本研究同时对比了Nanopore连接法和快速法测序效果,连接法的建库时间约4 h,测序速度快、深度更高,1 h可以满足分析需求;快速法的优点在于无需纯化,建库时间短(20 min),SARS-CoV-2全基因组序列最快2 h内获得,但5个样本测序深度总体不高,产生测序数据速度慢。实际工作中快速法更适合长片段序列,连接法适合短片段序列,这两种建库方法也为三代测序提供更多选择。
本研究中Nanopore三代测序的主要限制因素在于变异位点检测数目较Illumina二代测序少,以及部分插入缺失,相信随着三代测序的大规模推广应用,测序技术及纠错软件的不断优化,数据准确度的不足必将会得到弥补[16]。对16份样本的全基因组测序中,Nanopore三代测序凭借速度快、分型准确等优点,能够在疫情暴发的初期迅速鉴别新冠病毒;Illumina二代测序错误率低,测序数据更加可信,这两种测序技术从不同应用层面为疫情风险评估、流行病学监测及公共卫生决策提供技术保证。本研究也有一定的局限性,如样本量较少未能全面反应总体状况,结论的适用程度有待扩充等,但本研究可为后续新冠病毒全基因组测序工作开展提供参考。
利益冲突:无
引用本文格式:李东晓,李懿,朱琳,等.两种高通量测序平台应用于不同SARS-CoV-2变异株的对比研究[J].中国人兽共患病学报,2022,38(9):771-777.DOI:10.3969/j.issn.1002-2694.2022.00.122
