结球甘蓝无蜡粉亮叶性状dCAPS标记引物优化
2021-09-09王超付天姿张晓烜李秀乐
王超,付天姿,张晓烜,李秀乐
(东北农业大学园艺园林学院,哈尔滨 150030)
结球甘蓝(Brassica oleraceaL.var.capitataL.)简称甘蓝,是十字花科蔬菜中重要一种。航海大发现时代传播到世界各地,明清时期传入我国[1]。初莲香等1993年初次发现结球甘蓝无蜡粉亮绿型突变体[2],该种突变体甘蓝在生长阶段植被表面均无蜡粉覆盖,与普通结球甘蓝相比差异显著。Justus和Stoner等报道指出,小菜蛾和部分鳞翅目昆虫等害虫在芸薹属无蜡粉植株上具有产卵偏好性[3-4],但瓢虫成虫和卵数量有所增加[5],该害虫天敌在无蜡粉植株上移动速度快于有蜡粉植株,益虫捕食能力提高使无蜡粉亮绿植株对芸薹属常见害虫具有一定抗性[6]。Cole等研究发现,无蜡粉亮绿性状对植株本身而言,增加对水胁迫敏感性,这一现象导致植株体内分泌阻止昆虫取食化合物浓度升高[7]。此背景下,无蜡粉亮绿突变体研究对于抗虫性等优势甘蓝品种选育具有重要意义。
多种分子标记技术已运用于结球甘蓝无蜡粉亮绿性状和目的基因定位相关研究,董昕探究甘蓝显性蜡质缺失基因BoGL-3定位与候选基因,通过BSA法筛选标记,利用F2群体验证标记,将甘蓝蜡质缺失基因BoGL-3定位在8号染色体末端83 kb区间内[8]。刘东明对甘蓝蜡质缺失基因BoGL-4作精细定位,利用SSR标记精细定位,将目的基因BoGL-4定位在1号染色体170 kb区间内[9]。李景涛选用无蜡粉亮绿结球甘蓝10Q-961和普通有蜡粉结球甘蓝10Q-206为父母本,在对目的基因定位时,利用gSSR和InDel多态性引物筛选F2群体,最终试验结果表明,共显性标记Scaffold2324与突变体结球甘蓝10Q-961基因gwl遗传距离为6.5 cM[10]。
在现有分子标记手段中,单核苷酸多态性(Single nucleotide polymorphisms,SNP)与其他标记方法相比,优势在于其遗传稳定性高、所在位点极具代表性且分布广泛,已成为热门分子标记方法之一[11]。除此之外,SNP检测方法较多,虽然大部分检测方法存在成本过高或操作繁琐等缺陷,遗传育种领域存在一定局限性[12-13]。但CAPS标记(酶切扩增多态性,Cleaved amplified polymorphic sequences)和衍生CAPS(衍生酶切扩增多态性,Derived cleaved amplified polymorphic sequences,dCAPS)是可将SNP多态转化成可酶切扩增的有效分子标记方法[14]。已有报道中司文洁[15]、王彩芬[16]和雷天刚[17]等研究证明,CAPS标记具有对DNA用量低、操作方便、共显性明显和多态性好等优点,故在模式植物拟南芥[18]、大田作物小麦[19]和大麦[20]及水果蔬菜作物中应用广泛。dCAPS标记开发具有一定复杂性,且因限制性内切酶多样性,dCAPS引物设计变得极其耗时,Li等开发批量开发工具(http://223.65.208.206.8018/)可直接设计引物对,且提供引物错配等级[21],还可使用Neff等开发dCAPS Finder 2.0软件(http://helix.wustl.edu/dcaps/dcaps.htm)设 计dCAPS引 物[22],该方 法 使dCAPS引物开发更方便快捷。
综上,dCAPS标记自发明以来凭借其位点多、高效快捷等特点在分子研究方面应用广泛,例如基因定位、品种品系鉴定、图位克隆等方面。本试验开展结球甘蓝dCAPS分子标记辅助育种,利用新概念分子育种理论和技术体系,实现从经验育种到高效育种的转变,进而加快结球甘蓝新品种选育进程。
1 材料与方法
1.1 试验材料
结球甘蓝蜡质缺失突变材料“亮叶98-1030”,结球甘蓝蜡质正常的野生型材料“98-1030”,以及其有性杂交F1、F2代单株。材料表型如图1所示,植株表现为亮叶色、有光泽,叶片表面和茎均无白色或灰白色粉状蜡质覆盖,则为无蜡粉亮叶突变型植株;植株表现为灰绿色,叶片表面和茎上均有一层白色霜状蜡质,叶脉表现为白色或黄白色,则为有蜡质正常型植株。以上材料均由东北农业大学园艺园林学院甘蓝课题组提供,同年同一时期播种于温室。

图1 野生型98-1030及无蜡粉亮叶突变体98-1030对比Fig.1 Comparison of wild-type cabbage 98-1030 with bright-leaf mutant without wax powder
1.2 试验方法
1.2.1 田间试验
试验材料于2019年春播种于东北农业大学园艺实验站温室内,待两叶一心时分苗于单个营养钵中并编号;在苗龄3~4片真叶时,调查性状,根据表型性状分离比作卡方检验。将编号植株分别取0.2 g,保存于-80℃条件下,备用。
1.2.2 亲本基因重测序及候选SNPs挖掘
将结球甘蓝野生型98-1030和无蜡粉亮叶突变型98-1030构建DNA文库作全基因组重测序,并筛选测序结果,与参考基因组作比对(https://www.ncbi.nlm.nih.gov/genome/10901?genome_assembly_id=59537),利用比对结果检测和筛选SNPs并注释统计,以上部分由深圳华大基因股份有限公司完成。最后使用IGV软件将测序所得的亲本基因与参考基因组作比对,获取目标区间SNPs。
1.2.3 dCAPS引物的设计
基于筛选得到的20个SNP位点设计dCAPS引物:先利用dCAPS Finder 2.0(http://helix.wustl.edu/dcaps/dcaps.htm)设计dCAPS标记错配引物,再利用Premier 5.0设计dCAPS标记下游引物。错配引物设计原则为上下游引物在23~25 bp之间,错配碱基数为一个,PCR产物条带在150~400 bp之间。上述引物均由北京睿博兴科生物技术有限公司合成,以PAGE方式纯化。
1.2.4 dCAPS多态性引物筛选
dCAPS引物多态性筛选具体步骤为:
(1)采用改良CTAB法提取双亲以及F1的DNA,并利用设计好的dCAPS引物作PCR扩增;
(2)记录PCR产物电泳带型,以条带清晰、易于识别、可稳定扩增为筛选条件,使用对应限制性内切酶酶切,最后统计酶切产物条带情况,筛选目标引物。
2 结果与分析
2.1 结球甘蓝无蜡粉亮叶98-1030遗传模式分析
试验所选5个F1单株全部表现为显性野生性状,于2018年4月开始授粉,6月获得F2代,同年7月播种于温室,分别编号,8月统计表型。F2代材料表现型分为无蜡粉亮叶型和表面蜡质正常型两类,表型性状统计结果为,无蜡粉亮叶型和表面蜡质正常型比例接近1∶3.95,经卡方检验(x2=10.65>x20.05=3.841)与孟德尔3∶1分离比例相差较大。此现象可能是因为F2群体较小,且仅来自3株F1植株造成。本课题组牟香丽等试验结果证明结球甘蓝98-1030无蜡粉亮叶控制基因为一对隐性基因控制的质量性状[23],遵循孟德尔遗传定律。
2.2 DNA提取与检测
按照改良CTAB方法提取野生型98-1030、突变型无蜡粉亮叶98-1030、F1及F2群体基因组DNA,琼脂糖凝胶电泳检测DNA,DNA样品条带如图2所示,条带清洗、无降解、无拖尾,证明DNA样品提取质量较高,可开展下一步试验,将提取后DNA原液放置-20℃温度下保存,使用时稀释5倍作为工作液用于PCR扩增。
图2 主要材料DNA检测结果Fig.2 DNA test results of main materials
应注意的是,PCR产物和酶切产物检测方法不同。PCR产物检测步骤为,待PCR反应完成后,配置浓度为1.5%琼脂糖凝胶,以120 V电压电泳,时长20 min。PCR产物酶切电泳方法为:待酶切完成后,配置浓度为3%琼脂糖凝胶,以80 V电压电泳,时长1.5 h。
2.3 基于双亲转录组测序SNP分析
利用双亲全基因组重测序比对分析发现,一些可能影响基因功能的SNP分布情况如下:6 652个SNP参与密码子的提前终止;1 395个SNP将终止密码子替换为其他氨基酸残基,使该SNP所在DNA片段翻译出更长开放阅读框;还有2 442个SNP可改变基因组上的剪接供体及受体位点;9 325个SNP导致甲硫氨酸残基改变。最后根据测序结果注释信息在目标区间内筛选出20个SNPs筛选dCAPS引物。
2.4 dCAPS标记转化
根据亲本基因组重测序结果在目标区域内选择20个SNP位点,将其转化为dCAPS标记,得到20对可用于PCR扩增,扩增片段包含特异性酶切位点dCAPS引物,分别编号为DC01~DC20。
以引物DC01为例,设计方法和原理见图3,该引物所使用限制性内切酶AvaII识别序列为GGW⁃CC,序列中W为A和T简并碱基国际通用代码。

图3 dCAPS引物DC01设计Fig.3 Design of primer DC01 for dCAPS
所设计上下游引物序列及所对应限制性内切酶见表1,表中加粗斜体字母为错配碱基。
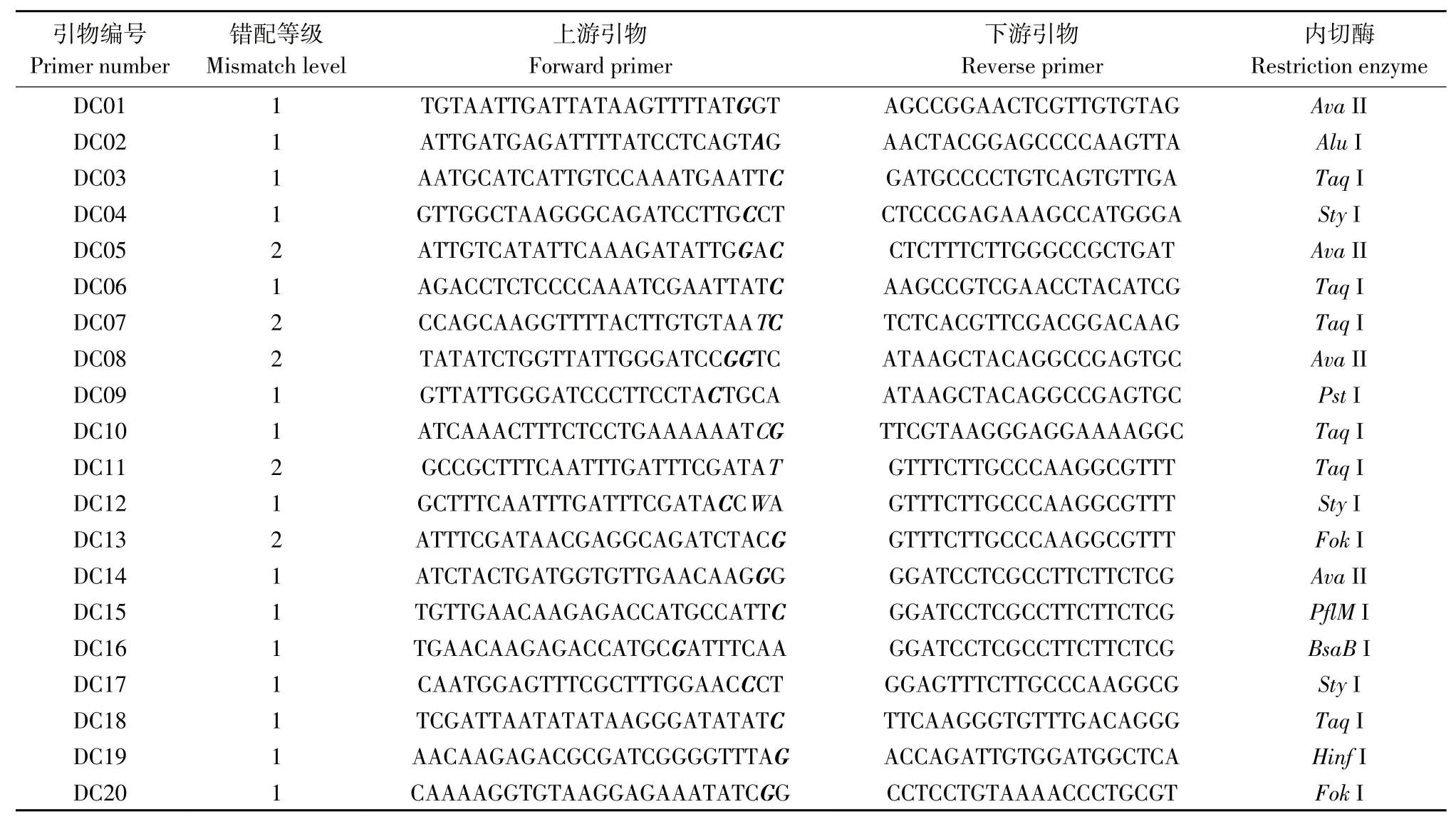
表1 根据SNP位点设计的dCAPS引物Table 1 dCAPS primers designed according to SNP
2.5 dCAPS标记筛选
将20对dCAPS引物开展野生型结球甘蓝98-1030和无蜡粉亮叶突变型结球甘蓝98-1030以及F1代PCR扩增及酶切。PCR结果表明,有10对引物可扩增出产物条带;酶切验证结果显示,有8对引物经限制性内切酶酶切后产生多态性。
图4为PCR扩增后产物条带情况概述。图4A中1号引物(ND07)和3号引物(ND12)为可开展后续酶切验证的引物;2号引物(ND08)扩增出多条条带,故舍去;4号引物(ND13)无清晰的扩增产物,故舍去。图4B中1号引物(ND06)可稳定扩增,2号引物(ND16)显性亲本无条带,故舍去。
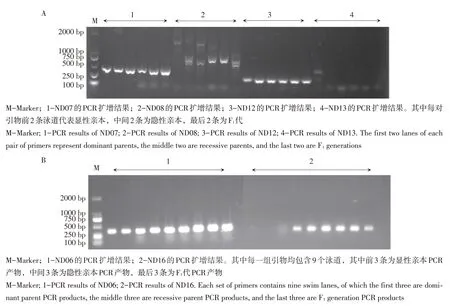
图4 部分dCAPS标记PCR扩增产物Fig.4 PCR products by dCAPS molecular markers
由于dCAPS标记中酶切位点一般包含于上游引物中,酶切产物即部分PCR产物被酶切得来,所以多态性差异较小,长度仅为20 bp。因酶切产物片段过于短小,在琼脂糖凝胶电泳中跑出凝胶区域,共显性酶切产物标记中一般仅存在2个条带片段,如图5所示。

图5 Sty I酶切产物检测结果Fig.5 Sty I digested product
酶切筛选后8对dCAPS引物为:DC03、DC04、DC06、DC07、DC11、DC12、DC16和DC19,转化效率为40%,可用于后续基因定位研究。
3 讨论
传统正向遗传学(Forward genetics)基因定位方法一般是构建遗传连锁图谱,但存在定位区间较大、定位精确性低等缺陷,且正向遗传学步骤过程繁琐,耗时较长[24]。近年来,随着第二代测序技术飞速发展,高通量测序技术逐渐成熟,测序成本降低,更多方便快捷的基因定位方法被开发。
SNP(Single nucleotide polymorphism)即单核苷酸多态性,是指基因中单个碱基发生转换、颠换等突变,共有4种突变形式:C-T(G-A)、T-A(A-T)、C-G(G-C)、C-A(G-T),但第一种突变形式频率最高,约占全部突变频率67%[25]。理论上来说,将CAPS和dCAPS相结合,可检测任意SNP位点[26]。但在SNP转化dCAP标记中,引物能否转化成功是关键,引物中错配碱基数以及错配位置距离引物3'端远近,均决定dCAPS标记开发能否成功。Neff等研究拟南芥探索试验中发现,当插入错配碱基在3'端第二位或者距离第二位更远位置时,可成功将限制性酶切位点引入引物中[22]。在PCR扩增时使用不同DNA聚合酶得到的PCR扩增结果可能会不同[26],PCR扩增时选用的DNA聚合酶不应具有校正功能,否则影响错配碱基插入,无法扩增PCR产物或者无法正常插入酶切位点。因本试验所选取SNP位点前后碱基均不存在酶切位点,无法直接设计得到CAPS引物,所以采用dCAPS标记方法筛选引物和优化。但部分dCAPS引物错配碱基距离3'端位置较近,可能导致部分PCR扩增效果差。本试验所选用DNA聚合酶为2×EsTaqMas⁃terMix酶,不具有高保真性,因此在DNA聚合时可保证顺利扩增及后续试验。
Di等研究玉米dCAPS标记开发检测时利用聚丙烯酰胺凝胶电泳法作酶切产物分离[27]。但聚丙烯酰胺凝胶相较于琼脂糖凝胶电泳而言,不仅制作过程复杂,且耗时久,故不推荐使用。可使用高分辨率琼脂糖凝胶检测酶切产物,Shahinnia等大麦dCAPS标记开发中利用高分辨率Metaphor琼脂糖凝胶[28];Yanagisawa等在小麦dCAPS分子标记中则利用另一种高分辨率Nusieve GTG琼脂糖凝胶[19]。与本试验所用普通琼脂糖凝胶相比,高分辨率琼脂糖虽效果较好,但价格过于昂贵,标记开发成本升高。
本试验所建立SNP/dCAPS转化体系基因定位方法在甘蓝作物中鲜有记载,本研究增加普通琼脂糖凝胶电泳浓度,利用降低电泳电压,延长电泳时间方法对差异较小酶切产物分离片段,达到经济实惠、操作简单且有效的目的。
4 结论
本试验根据野生型结球甘蓝98-1030和突变型98-1030的基因组重测序数据所得的非同义SNP位点作为基础,设计20对dCAPS引物,利用双亲及F1代作引物筛选及优化,最终筛选出8对PCR产物清晰、酶切后可产生特异性片段的共显性dCAPS标记:DC03、DC04、DC06、DC07、DC11、DC12、DC16、DC19,标记转化率为40%。研究结果可为后续基因定位及结球甘蓝优良品种选育奠定理论基础。
