LncRNA的靶RNA 预测研究
2021-09-07郑希强宋子健李建伟
郑希强,宋子健,李建伟
(河北工业大学人工智能与数据科学学院计算医学研究所,天津 300401)
随着生命科学研究的深入和高通量测序技术的飞速发展,研究发现大多数人类基因组均已转录[1]。据报道[2],其中仅有2%的RNA 分子可翻译为蛋白质,绝大部分RNA 是不能编码蛋白的非编码RNA。其中,长度超过200 个核苷酸的非编码RNA被定义为长链非编码RNAs(long non-coding RNAs,lncRNAs)。近年来,越来越多的研究表明,lncRNAs 参与调控多种重要的生物过程,如调控表观遗传染色质修饰、转录调控和转录后调控等[3]。2012 年,Gong ZJ 等[4]发现HOTAIR 可通过与PRC2复合物相互作用,诱导H3K27 发生甲基化,进而抑制该基因的表达。此外,lncRNAs 与癌症[5]、心血管疾病[6]和糖尿病[7]等多种复杂疾病的发生发展均存在着密切联系。因此,深入了解lncRNA的调控功能,对人类复杂疾病的诊断、治疗和预后提供新的治疗靶点具有重要意义。
1 LncRNA的靶RNA 预测研究
迄今为止,海量的lncRNAs 被不断发现,但对其调控功能的研究仍不够充分。通过传统的生物实验验证lncRNA调控功能,结果虽然准确、可靠,但存在实验周期长、费用高、效率低等问题。单纯地依靠传统的生物实验已远远不能满足当前需求,借助高效的、基于生物信息学的计算方法预测lncRNAs的调控功能已成为计算医学研究和对复杂疾病机制研究的热门方向。研究发现[8],lncRNAs 常通过碱基互补配对的方式调控靶RNA的表达,进而发挥其调控作用。因此,根据lncRNA 与靶RNA 之间的相互作用关系,通过计算预测lncRNA的靶RNA,利用靶RNA的生物学功能推断lncRNAs的调控功能已成为一种常见的研究方法。此外,lncRNAs 会折叠成复杂的二级结构来维持它们的稳定性和保守性。lncRNA的二级结构可以帮助预测lncRNAs的靶RNA[9]。lncRNA 二级结构的生物学意义及其碱基序列组成同样重要,部分lncRNAs的功能活性通常由它们特定的二级结构决定。
基于以上分析,开发lncRNA 靶RNA 预测模型在全基因组范围预测lncRNA-RNA 相互作用,快速搜索lncRNA 与RNA 之间最稳定的碱基互补配对状态,成为lncRNA-RNA 相互作用预测的关键[10]。随着生物信息学的不断发展,涌现出了数量众多的RNA-RNA 相互作用预测模型,这些模型均可用于lncRNA 靶RNA 预测。常见的RNA-RNA 相互作用预测模型见表1。

表1 常见预测lncRNA 靶RNA 模型的关键特性
2 常见的RNA-RNA 相互作用预测模型
2.1 LncTar LncTar 预测模型[11]通过标准化自由能(ndG)辅助科研人员预测lncRNAs的靶RNA。Li J等[11]从RNA 碱基互补配对的角度分析了lncRNAs与靶RNA 之间的相互作用关系,并将多重PCR 技术中引物二聚体的思想运用到筛选lncRNA 靶RNA中。LncTar 预测模型利用“滑动”算法和最近邻能量模型[15,16]寻找并计算结合区域的自由能,最终通过计算ndG 与自定义阈值实现lncRNA-靶RNA 相互作用关系预测。
LncTar 预测模型包含三个步骤。首先,创建二维矩阵,用于记录两个RNAs 分子的互补碱基对。在计算两个RNAs 形成的结合区域时,采用最近邻能量模型。其中自由能(ΔG)由给定的焓值(ΔH)、熵值(ΔS)和特定温度下的熔化温度T 来计算。具体计算公式:ΔG=ΔH-TΔS。其次,遍历两个输入RNAs 分子形成的全部结合区域,分别计算它们的总自由能(dG),并记录在哈希表中。遍历完成后,在哈希表中寻找最小的总自由能(dG)并获得其结合区域。最后,LncTar 提出一个判定标准——标准化自由能(ndG),用它来反映自由能最小的结合区域中lncRNA-靶RNA 结合的相对稳定性。具体计算公式如下:

其中结合区域长度(bindingregion)是两个RNAs序列中较短的RNA 序列长度。如果计算出两个RNAs 形成结合区域的ndG 小于等于人为自定义阈值,就可以判定两个RNAs 分子有相互作用关系,否则给出相反判定。
LncTar 预测模型的优势在于对RNA 序列长度没有任何限制,可适用任意长度的RNA 分子。并且LncTar 提供了标准化自由能,能自动判定两个RNAs 分子是否有相互作用关系,极大地减轻用户的判定负担。此外,LncTar的算法时间复杂度仅为O(n2)。但LncTar 预测模型也有一定的局限性。如因为追求计算速度,LncTar 没考虑到RNA 二级结构,而lncRNA的二级结构对大部分lncRNA-靶RNA相互作用的判定有着一定的影响。此外,如果两个RNAs 转录本序列长度较长的情况下,预测出的结合区域长度过长,将导致ndG 值过小,具有靶向关系的两个RNAs 会被误判。而ndG的阈值如果设置过小,虽然降低了假阳性率,但会导致一部分正样本预测结果不准。因此,LncTar 预测模型还有很大的提升空间。
2.2 RIsearch2 RIsearch2 模型[12]适用于大规模预测RNAs 间的相互作用。Ferhat A 等[12]在允许G-U 摇摆配对的情况下搜索与种子反向互补匹配的序列片段,并使用后缀数组索引与动态规划扩展种子的方法快速定位查询序列和目标序列之间的互补结合区域。RIsearch2 模型分为两个阶段。第一阶段:将查询序列和目标序列各自转换为后缀数组索引结构,并存储目标序列后缀数组的反向互补序列。转换为后缀数组索引结构的算法是基于GUUGle 算法[17]。大部分算法将Watson-Crick 对(A-T,C-G)视为匹配,并且允许存在G-U 摇摆配对。而Ferhat A 等提出一种无需反转查询序列来找寻结合区域的方法,将GA 和U-C 匹配视为有效对,然后使用二分查找来确定目标序列后缀数组中与查询序列匹配的区域。这种方法可以直接匹配目标序列的后缀数组,不需要反转查询序列。第二阶段:为了减少种子相互重叠造成的冗余,首先将冗余的匹配种子进行过滤,然后通过填充种子上下游区域的动态规划矩阵扩展剩余的种子区域,接着利用RIsearch[18]中简化的最近邻能量模型计算种子匹配区域的自由能,并与用户定义的分子间自由能阈值进行比较,筛选出满足条件的匹配区域。最后,通过回溯动态规划矩阵找到实际的相互作用区域。
RIsearch2 预测模型将RIsearch 模型与GUUGle模型进行深度集成,并且采用多个线程并行处理(OpenMP API)的方式对两个后缀数组进行匹配。这种高效的计算方式不仅克服了传统算法一次只能处理一个目标序列的障碍,还提高了计算速度。但RNA-RNA 相互作用预测中忽略RNA 二级结构信息会降低预测的准确性。
2.3 RIblast RIblast 预测模型[13]一种基于种子扩展方法[19]的超快速RNA-RNA 相互作用预测模型。首先,RIblast 使用后缀数组寻找种子匹配区域;其次,通过互补能量模型扩展种子区域的两端;最后,RIblast 模型使用两种能量之和作为目标序列和查询序列相互作用结合区域的输出结果。其中一种为可访问能量,这种能量是防止结合区域内形成分子内碱基对所需的能量,通过使用分区函数算法进行计算[20]。另一种能量为杂交能量,它源自两个RNAs 片段之间分子间碱基配对产生的自由能,可以基于最近邻能量模型计算。
RIblast 预测模型分为两个步骤:数据库构建和RNAs 结合区域搜索。在数据库构建中,首先是计算目标RNA 数据集中每个片段的可访问能量,然后将目标RNA 序列反转,并在未反转与反转的序列之间插入分隔符,接着构造串联序列的后缀数组索引,为了加快RNA 相互作用区域的搜索,可对短字符串预先计算其杂交能量,最后将可访问能量串联序列的后缀数组索引和短字符串计算互补能量的结果存储在数据库中。在RNAs 结合区域搜索中,首先是计算查询序列的可访问能量,并为查询序列构建后缀数组索引,然后基于查询和数据库的两个后缀数组,查找杂交能量小于阈值能量的种子区域。接着,通过杂交能量和两个可访问能量的总和来计算找到的种子区域的相互作用能。继次,RIblast 在种子区域两边进行扩展,没有间隙。当扩展中不再形成RNA 互补双链体时,RIblast 终止扩展。最后,删除与其他种子区域完全重叠的种子。另外,也排除超过阈值能量的相互作用结果。
对于RNA-RNA 相互作用预测,可访问能量的计算时间与序列长度成线性关系,杂交能量的计算时间是种子匹配项的平方,这是提高运行速度的障碍。而RIblast 先进行了大量的预处理工作,然后在查询序列和目标序列之间找到短匹配区域以加快速度(称为种子),最后将检测到的种子两端扩展,这种方法解决了时间复杂度高的问题。RIblast的不足之处在于最后只给出能量信息,部分用户无法通过这些能量信息判断两个RNAs 分子是否存在相互作用关系。没有给予用户一些判定标准,增加了人工判定的负担。
2.4 ASSA Antonov I 等[14]发现总自由能的值取决于RNA的转录本长度和GC 含量两个因素,这使研究人员很难比较不同特性转录本所计算出的RNARNA 相互作用的总自由能。为了解决这个问题,ASSA 预测模型[14]根据两个相互作用RNAs的转录本长度和GC 含量设计出了总自由能的统计显著性(P值)。ASSA 预测模型包括三个步骤。第一步,使用LASTAL[21]程序包中的局部序列比对工具,搜索查询序列和目标序列中的局部互补区域。在这项工作中,ASSA 将这些局部互补区域称为“反义位点”。第二步,首先提取LASTAL 预测的反义位点以及侧翼序列组成“假定双链体”,然后根据热力学模型计算其杂交能量。“假定双链体”中可能存在局部的RNA 二级结构,研究人员通过观察总结出两种“假定双链体”类型。第一种类型称之为“真双链体”,这种类型的RNA 双链体与任一侧附近的(侧翼)区域不具有强互补性,即单链RNA 无法形成二级结构。因此,这种类型的RNA 双链体只形成分子间碱基对。第二种类型称之为“两个发夹”。除了RNAs 分子间碱基互补配对以外,两个RNAs 自身还具有完美的(100%)分子内互补性。为了将“真双链体”与“两个发夹”两种类型区分开,ASSA 预测模型使用基于热力学的工具RNAup[22],因为它能够通过计算分子间自由能相对准确地分开两种类型。ASSA 将RNAup 应用于特定序列组块(“假定双链体”)而不是全长转录本,这样可以有效并快速地计算RNA 分子间的杂交能量。最后,全部特定序列组块相加当作两个RNAs 结合产生的总自由能。第三步,估算获得的总自由能的统计显著性。通过将观察的RNA 序列[总自由能的分布取决于RNA 序列的特征(长度和GC 含量)]与相应随机序列(ASSA 生成的负样本)产生的总自由能进行比较,估算总自由能的统计显著性P值。具体计算公式如下:
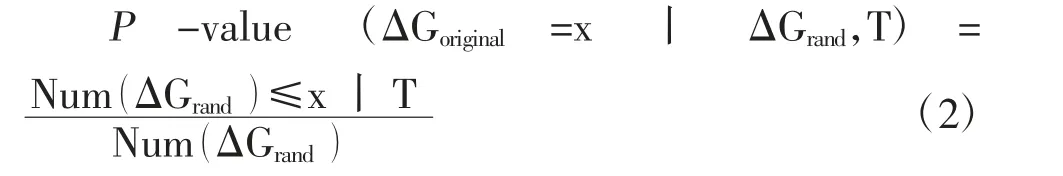
其中x 是观察的RNA 序列通过ASSA 预测的总自由能,Num(ΔGrand≤x|T)是随机序列产生的总自由能小于或等于x的随机序列个数,Num(ΔGrand)是随机序列的总数。统计显著性P值越小,说明观察的两个RNAs 有相互作用的可能性越大。
在ASSA 中的主要创新是数学统计模型,它可以快速计算并估计观察的RNA-RNA 相互作用产生总自由能的统计显著性。在RNA-RNA 相互作用预测模型中,ASSA 是唯一估计P值的预测模型。由于仅使用局部RNA 二级结构,对于全序列来说,反义位点上RNA 二级结构可能超出它侧翼区域的范围,忽略该二级结构的自由能会对最后结果的准确性产生一定的影响。
3 总结
不同的lncRNA 靶RNA 预测模型各有其自身的特点和优势。研究者应根据研究目的和需求,进行合理选择。但目前lncRNA 靶RNA 预测模型还存在一些不足之处需要深入研究,主要包括以下几个方面:首先,由于lncRNAs的研究刚刚起步,相关的lncRNA 生物特性认识仍然不足。部分lncRNAs的生物特征如何表征还需进一步探索,如RNA的三级结构,其空间性特征可能在预测lncRNA 靶RNA 中发挥重要的作用。其次,实验验证的数据虽然可靠,但数据量较少,通过计算统计的方法来区分两个RNAs 相互作用的规律还需要进一步研究,以提高预测模型的灵敏度。最后,还应考虑其他因素,包括RNA的染色体定位以及特定RNA 结合蛋白的存在。发现和引入RNA的更多特征是lncRNA 靶基因预测模型发展的一个重要方向。
随着更多相关数据库地建立和RNA-RNA 预测方法的改进,相信会有更多、更准确的lncRNARNA 相互作用预测模型被开发并应用于lncRNA调控功能的研究。总之,在全基因组水平上预测lncRNA-RNA 相互作用的关系来寻找lncRNA 靶RNA 仍然有巨大的进步空间。
