磷酸根修饰的Mn 掺杂介孔TiO2 在VUV-PCO 体系高效催化氧化甲苯性能
2021-09-01舒亚婕梁诗敏肖家勇涂志凌黄海保
舒亚婕,梁诗敏,肖家勇,涂志凌,黄海保,*
1 中山大学环境科学与工程学院,广州 510274
2 珠海市金湾区联港基础投资有限公司,广东 珠海 519000
3 湖南建工集团有限公司,广东 珠海 519000
1 前言
近十年来,我国大范围、大规模复合型大气污染状况频发,而挥发性有机污染物(VOCs)成分复杂,不仅是大气环境气溶胶颗粒物(如PM2.5)、雾霾和光化学烟雾等污染的关键前驱体,还对大气环境、动植物及人类健康造成严重威胁1,2。其中苯、甲苯等典型芳香族化合物毒害作用较大,具有致畸致癌致突变效应。尽管各国陆续出台多项政策对VOCs进行控制和治理3,但大部分成熟的治理方法如吸收法4、催化燃烧技术5、生物法6、等离子体技术7等主要针对中高浓度气态污染物。工业、交通、农业等过程排放的废气通常浓度较低且风量体量大8,采用传统方法处理存在投资和运行成本高9以及潜在爆炸危险等不足,因此急需开发出高效、低成本、安全的,针对低浓度气态污染物的处理技术,来满足社会的实际需求。
光解催化氧化技术能够避免高温热力焚烧或催化燃烧等存在的诸多局限性,具有效率高、反应条件温和、过程简单以及节能环保等优点,是治理低浓度气态污染物的新技术,极具未来发展趋势10,11。真空紫外灯由于其结构、价格与传统254 nm杀菌灯相差无多,但却能发射约占总能量8%的185 nm紫外光,效率高、成本低廉,因此真空紫外灯可直接用于光降解气态污染物12。185 nm紫外光不仅能直接光解气态污染物,还能激活空气中的水蒸气和氧气,产生∙OH、∙O和O3等活性氧物种间接氧化气态污染物13。同时VUV真空紫外灯发射的185和254 nm紫外光均可被光催化剂吸收,在催化剂表面发生光催化反应,产生大量强氧化性物种,用于进一步氧化14,15。Chen和An等16对比了气态苯乙烯在254和185 nm照射下TiO2光催化降解机理,发现VUV光解H2O和O2可大大增加∙OH,有利于性能的提升。因此将VUV光解技术及光催化技术(PCO)相结合,可以大大提高光能利用率,同时VUV光解产生的中间产物可进一步催化氧化成无毒无害的CO2和H2O,减少二次污染。此外,臭氧同样也是VUV技术光解产生的毒副产物,但是臭氧本身具有极强的氧化性17,18,因此在消除臭氧的同时可以在催化剂的辅助下将其转化利用,发挥体系中VUV光解、光催化氧化和臭氧催化氧化协同作用的优势。
商业二氧化钛(P25)由于自身较大的禁带宽度(3.2 eV)和高电子空穴对复合率,其降解和利用臭氧的效率不高19。多级孔结构或者材料中含有大孔、介孔和微孔等发达的各类孔道,有利于降低催化剂本身的阻力,加速物质传输传质,有效地把物质传送到催化剂活性位点,加快反应速率,增强其催化性能,因此在气态光催化反应中更具优势20,21。研究发现22,过渡金属氧化物,特别是锰氧化物23,表面上暴露有未饱和的原子或氧空位,具有较高的臭氧分解性能。因此,为了提高催化剂对VOCs的吸附性能,抑制光生载流子复合,提高光催化效率和臭氧分解能力,我们对介孔二氧化钛进行改性,磷酸修饰和Mn掺杂共同修饰,使其在VUV光照下既能高效分解利用臭氧,又能吸收光能有效光催化氧化降解气态污染物。TiO2表面吸附如F−和PO43−等阴离子,有助于吸附O2等氧物种,从而有效吸引和捕获电子,提高光生载流子分离效率,从而增强光催化氧化性能24,25。此外,研究发现26,F−和PO43−等阴离子可取代表面的羟基基团,有助于生成未束缚的流动态羟基自由基(而非表面羟基),从而高效降解污染物。
因此本文以甲苯作为目标污染物,采用真空紫外灯,通过制备磷酸和Mn共同修饰的介孔二氧化钛(P-Mn-TiO2),使其具有光催化与臭氧催化氧化活性,在甲苯被真空紫外光解的同时,利用光解副产物臭氧与紫外光源进行臭氧催化氧化以及光催化,在一个反应装置内实现真空紫外光解(VUV光解)、光催化(UV-PCO)以及臭氧催化氧化(OZCO)三种功效的协同。本实验拟通过SEM、TEM、UV-Vis、XRD、XPS等表征手段分析催化剂结构特征与活性的构效关系,探究P、Mn改性对复合催化剂的光催化活性、臭氧催化活性以及吸附性能的影响机制。并探究其协同机理,从而在理论上更好的指导技术的开发与优化。
2 材料与方法
2.1 实验装置
本文采用自制连续流动式反应装置,VUVPCO工艺流程图如图1所示,主要包括配气系统、VUV光反应系统和检测系统。其配气系统主要是通过装有碱石灰和变色吸水硅胶的气体净化器(GPI-2,福立仪器设备有限公司)净化空气发生器(HGA-2L,北京汇龙昌海科贸公司)中产生的空气,以获得不含有水、CO2及有机杂质的零气作为系统的气源。通过鼓泡法,经质量流量控制器(S49,北京汇博隆精密仪器有限公司)分别控制污染物甲苯(色谱纯,Mreda Technology,Inc.,美国)、水、干空的流量,获得甲苯蒸气和水蒸气,经过缓冲瓶充分混匀,配置成一定浓度、湿度及流量的有机气体;在本体系中总流速为1 L·min−1,甲苯浓度为40 ppm(1 ppm = 10−6)。如没有特别指出,湿度为50%左右。VUV-PCO光反应系统由包含真空紫外灯及负载催化剂两部分。其反应器有效容积约1.5 L,2根4 W的185/254 nm紫外灯(SSUV-A02,佛山市君睿光电科技有限公司)垂直放置于反应器两侧,将装有催化剂的内径为0.8 cm的砂芯层析柱构成石英玻璃管放入在两根灯的中间。混合气体由反应器顶部进入,从石英管排出。催化剂为0.5 g,空速为180000 mL·g−1·h−1。反应后的气体,被分为3路,分别进入气相色谱仪(GC-9790 plus,福立)、臭氧检测仪(Modle 106-L,美国2B Technology)和净化排空。其中,气相色谱仪测定反应系统出气中气态甲苯和CO2浓度。
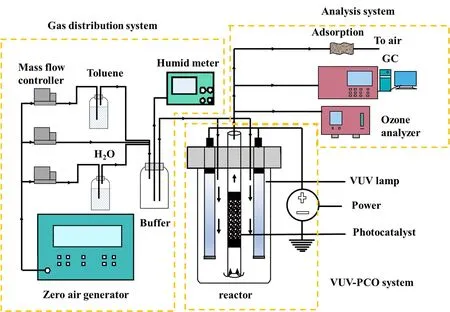
图1 VUV-PCO系统示意图Fig. 1 Schematic diagram of VUV-PCO system.
2.2 催化剂的制备
(1) 一步水解法制备介孔TiO2催化剂,选用钛酸四丁酯(AR,天津福晨化学试剂厂)为前驱体。在烧杯中量取200 mL二次蒸馏水;不搅拌的情况下,向密封罐中滴入20 mL钛酸四丁酯;静置老化24 h;过滤白色沉淀物,并用蒸馏水反复洗涤多遍;真空50 °C干燥12 h后400 °C下煅烧1 h,得到的产物记为TiO2。
(2) 采用浸渍法制备P-Mn-TiO2催化剂。根据计算所需的负载量,上述200 mL蒸馏水的烧杯中溶解相应质量的乙酸锰前驱体,搅拌半小时,至溶解;不搅拌的情况下,向密封罐中滴入20 mL钛酸四丁酯;静置老化24 h;过滤白色沉淀物,并用蒸馏水反复洗涤多遍;真空50 °C干燥12 h后400 °C下煅烧1 h,得到Mn-TiO2。随后将Mn-TiO2浸渍于一定浓度的磷酸溶液中(AR,阿拉丁),于80 °C下搅拌12 h,洗涤、过滤,80 °C干燥后,300 °C焙烧1 h,得到P-Mn-TiO2。
实验前将所有催化剂在10 MPa下压成片状,经研磨后筛分,取20-40目颗粒作为实验用催化剂。
2.3 表征方法
本实验采用冷场发射扫描电镜(SEM)(JSM-6330F,日本电子株式会社)、透射电镜(TEM) (FEI Tecnai G2 Spirit,日本)分析催化剂的形貌;利用BET (Brunauer-Emmett-Teller,BET,美国)方程计算催化剂的比表面积,利用等温线吸附分支并采用BJH (Barrett-Joyner-Halenda,BJH)模型计算介孔材料的孔容和孔径分布。采用紫外可见光吸收光谱(UV-600,日本岛津)评价催化剂的光响应性能。催化剂的晶体结构采用荷兰帕纳科Empyrean型X射线衍射仪(XRD)进行分析。采用铜靶(CuKα辐射,λ= 0.154056 nm),电压40 kV,靶电流25 mA,以4 (°)·min−1的速度连续扫描。催化剂上纳米晶体尺寸采用Scherrer公式估算,但只有当粒径小于或等于100 nm时,计算值才与实际值相近。通过X射线光电子能谱仪(Thermo-ESCALAB 250XI,Thermo Fisher Scientific,美国)测定催化剂的光电子能谱图,分析样品(除H原子外)表面的元素组成及其化学环境和价态信息。X射线光源是单色AlKα(1484.6 eV)源,工作电压为15 kV,发射电流50 mA,束斑大小为500 μm。实验测定的各元素的结合能(BE)通过以表面污染C 1s(284.8 eV)为标准进行校正。
3 实验结果与讨论
3.1 复合催化剂表征分析
3.1.1 复合催化剂结构和形貌分析
图2为不同催化剂:未掺杂的介孔TiO2、Mn掺杂的介孔Mn-TiO2和磷酸修饰的P-Mn-TiO2的XRD衍射谱图。从图中可以看出,所有样品在2θ= 25.2°时出现了明显的对应于锐钛矿TiO2的101晶面的衍射峰(JCPDS 21-1272),没有金红石相特征峰27,从而说明合成的介孔二氧化钛为纯锐钛矿型,表面修饰并未对其本身的晶型结构产生影响。此外,TiO2、Mn-TiO2以及P-Mn-TiO2的衍射峰强度和峰型相似,这可能是由于合成的是介孔结构的二氧化钛,其结晶度较低,晶粒尺寸小。此外,尽管合成过程中掺杂了Mn元素,但Mn-TiO2和P-Mn-TiO2的晶型和结构并未发生变化。值得注意的是,在Mn-TiO2的衍射图谱中,并未检测到明显的Mn2O3或Mn3O4的特征峰,这可能是由于Mn的负载量较低,或者是因为Ti4+和Mn4+/Mn3+有相似的离子半径,Mn替换Ti掺杂入TiO2晶格中28。
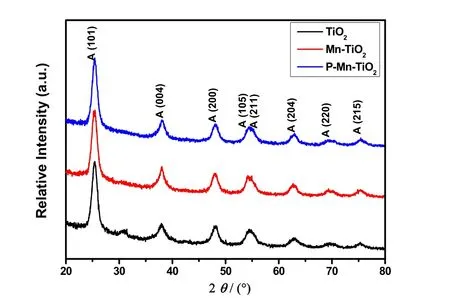
图2 不同催化剂(TiO2、Mn-TiO2和P-Mn-TiO2)的XRD图Fig. 2 XRD patterns of different samples(TiO2, Mn-TiO2和P-Mn-TiO2).
为了更清晰地了解催化剂的形貌、表面性质和结构,我们对P-Mn-TiO2进行扫描电镜、透射电镜(TEM)以及高倍透射电镜TEM的分析,我们可以从图3中清晰的看到,P-Mn-TiO2是以均一的纳米晶粒构成的微球,其晶粒尺寸较为均一,为8-10 nm。微球尺寸为1-2 μm,有部分坍塌现象,表面不光滑,这可能是由于磷酸处理所致,说明P-Mn-TiO2是由均匀的介孔微球堆积成的具有多级孔结构的材料。从高倍电镜TEM分析中可以清晰的看到P-Mn-TiO2的微结构和晶格条纹,其只含有0.35 nm晶面间距,对应于锐钛矿二氧化钛的(101)晶面(JCPDS card No. 89-4921),和XRD结果一致,即PMn-TiO2仅为锐钛矿型。

图3 P-Mn-TiO2微球的(a)扫描电镜图、(b, c)透射电镜图和(d)高倍透射电镜Fig. 3 (a) SEM images; (b, c) TEM images and(d) HRTEM images of P-Mn-TiO2 beads.
3.1.2 比表面积及氮气吸附脱附性能
表1和图4所示为不同材料TiO2、Mn-TiO2和PMn-TiO2的表面织构性质、氮气吸附脱附曲线和Barrett-Joyner-Halenda (BJH)孔径分布。根据IUPAC分类,三个样品的氮气吸附-脱附等温曲线均呈现为IV型等温线,在相对高压区有H1型滞回环29,30,说明TiO2、Mn-TiO2和P-Mn-TiO2均为介孔材料,孔径为2-50 nm间。相对于含有整齐均一的介孔TiO2,Mn-TiO2的N2吸附量上升,其滞回环向P/P0= 1偏移,这可能是由于其介孔变大所致,而P-Mn-TiO2的N2吸附量则相对于Mn-TiO2出现下降,则可能是由于部分孔道坍塌。小图所示为不同材料的孔径分布图,TiO2的孔径尺寸约为5 nm,而Mn-TiO2和P-Mn-TiO2的孔径要略大于TiO2的,以6 nm左右为主。表1列出了催化剂的孔直径、孔体积和BET比表面积,其中Mn-TiO2的BET比表面积相对较高,为153.2 m2·g−1,孔体积为0.354 cm3·g−1。当磷酸修饰后,催化剂P-Mn-TiO2的比表面积略微下降为130.6 m2·g−1,其孔体积为0.278 cm3·g−1。说明Mn的加入改善介孔二氧化钛的孔道结构,但是磷酸修饰后可能影响催化剂的规整介孔大孔的生成,或是形成的颗粒覆盖或堵塞部分孔道。
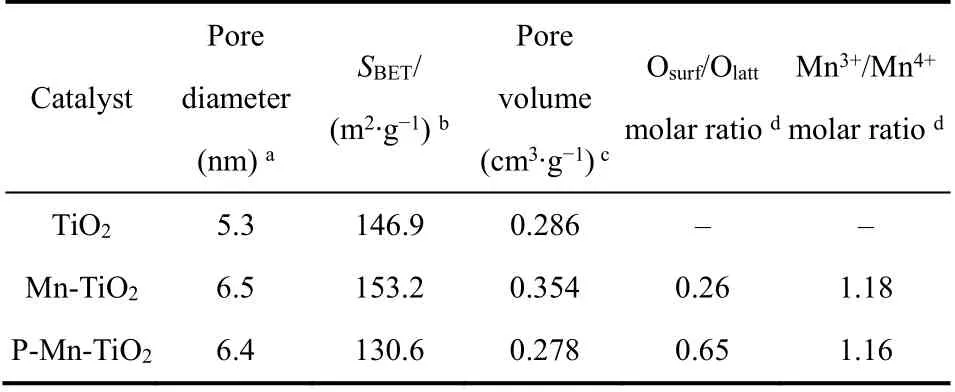
表1 不同光催化剂的表面结构特征Table 1 Textural properties of TiO2, Mn-TiO2 and P-Mn-TiO2 catalysts.
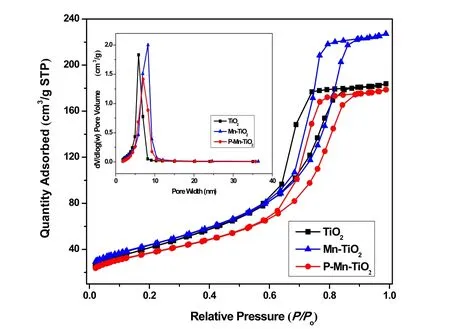
图4 不同催化剂TiO2,Mn-TiO2和P-Mn-TiO2的氮气吸附-脱附等温曲线及孔径分布图Fig. 4 Nitrogen adsorption-desorption isotherms and the corresponding pore size distribution plots (inset) of the TiO2, Mn-TiO2 and P-Mn-TiO2 catalysts.
3.1.3 XPS分析
为了获得更深入的催化剂表面性质,确定P和Mn的存在状态,我们对P-Mn-TiO2进行了X射线光电子能谱(XPS)测试,其中,所有的谱线均通过C 1s峰结合能为284.6 eV进行校正。图5a所示为合成的Mn-TiO2和P-Mn-TiO2的Ti 2p的光电子能谱图,从图中可以看出有两个对称的自旋轨道双峰(Ti 2p3/2,结合能为458.8 eV;Ti 2p1/2,结合能为464.6 eV)31,均代表锐钛矿TiO2的Ti4+。我们并未在谱图中检测到Ti的多价态或是还原Ti2+,说明P、Mn修饰不会影响到TiO2中Ti原子的本身化学性质。在O 1s的XPS谱图中,可以将谱图分峰成为3个峰32:O2−,晶格氧Olatt;O22−表面活性氧物种Osurf;和―OH羟基物种和表面吸附水(532.5 eV),从图中分析看到Mn-TiO2催化剂的晶格氧Olatt(529.9 eV)特征峰更显著,这可能是由于体系中存在MnOx所致。而P-Mn-TiO2催化剂的Osurf物种要显著高于Mn-TiO2,从表1中可看出其Osurf/Osurf摩尔比为0.65,是Mn-TiO2催化剂的2.5倍,进一步这说明磷酸修饰后催化剂表面有更多的表面吸附氧位点或氧空位,有益于催化反应的进行。
为了明确Mn的离子状态,图5c显示了Mn-TiO2和P-Mn-TiO2表面的Mn 2p轨道精细谱图。从图中看出,Mn呈现出宽而不对称峰形,从而说明该催化剂的Mn元素是呈现混合价态。对Mn 2p3/2谱图进行分峰,可将其分为结合能为641.0-642.5 eV和643.3-644.5 eV两个特征峰33,其中一个主峰在641.3 eV 代表Mn2+或Mn3+的存在(641.3-641.9 eV)。另一个是有更高结合能的643.1 eV,代表着Mn4+的存在。由于Mn2+和Mn3+的结合能相近(641 eV左右),因此在本材料中很难完全确定是Mn2+或Mn3+的存在。但是,大量研究认为34,35,一旦Mn3+出现在锰氧化物中,因为静电平衡的原因,催化剂表面氧空位就会随即产生。磷酸处理后P-Mn-TiO2的Mn3+的含量下降,说明Mn原子的氧化状态可能升高,其价态从低价+3部分氧化为+4。从P 2p的结合能图谱中可以看出,可以清晰的看到结合能在133.5 eV所代表为磷酸吸附在TiO2的表面36。综上发现,磷酸以吸附的形式修饰在Mn-TiO2表面,并未改变催化剂的晶型结构和晶相组成,仅使催化剂表面具有更多表面活性氧物种,有益于接触催化反应,提高催化剂表面光生载流子分离性能。
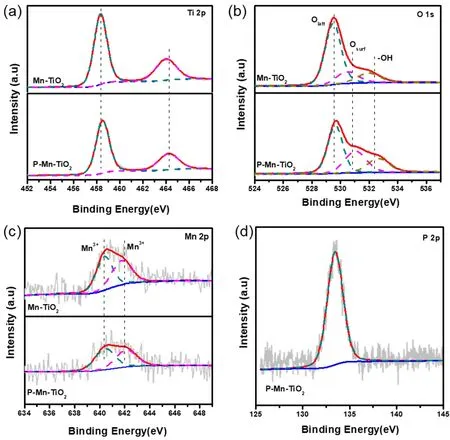
图5 不同催化剂的XPS图谱(a) Ti 2p;(b) O 1s;(c) Mn 2p;(d) P 2pFig. 5 XPS patterns of (a) Ti 2p; (b) O 1s; (c) Mn 2p; (d) P 2p.
3.1.4 紫外可见光吸收性质(UV-Vis)
UV-Vis吸收光谱可分析光催化剂的吸光能力及光吸收边带的移动情况,而不同晶体结构和能带结构的光催化剂,其表现出的光学性质也不一样。如图6所示为不同催化剂的UV-Vis吸收光谱图。从图中可以看出,P25和介孔TiO2的光吸收边带范围一致,均为400 nm范围内,这是由半导体材料电子从价带激发到导带的本身固有性质所决定,说明P25和介孔TiO2仅有紫外光响应。其中,TiO2的吸收光强更高,表明介孔结构的纯锐钛矿二氧化钛具有更高的UV光吸收能力。而Mn的掺杂使二氧化钛在可见光范围有明显的提高,从而扩宽了催化剂的吸光范围,从紫外到可见光均能使催化剂产生光生载流子,从而有利于提高光催化反应的应用范围。此外,Mn-TiO2能对可见光有响应说明其带宽减小并出现多个中间带隙转化现象37。通过UV-Vis图谱转化为图6b可知,Mn-TiO2和P-Mn-TiO2样品的禁带宽度减小,都为1.8 eV。由于Mn和Ti之间的相互作用,Mn掺杂可能会在TiO2的带隙中产生多个占有电子态。而带宽则是由于半导体受光激发后产生的光生电子从体相到表相的转移所引起的。Mn3+/Mn4+离子掺杂进TiO2的晶格内会出现d轨道分裂,从而产生可见光响应。同时由于Mn3+/Mn4+的掺杂,其能级低于TiO2的导带底,为了能够维持电荷平衡和中性,氧空位随即产生。这个结果与最近的理论研究结果34,38相一致,即通过Mn3+/Mn4+离子取代Ti4+离子进入到TiO2晶格中,无论是从总体上减小带隙,还是从禁带中引入中间带,都会导致TiO2发生显著的红移。根据理论计算,这些带隙之间的中间能级可能是TiO2晶格中过渡金属Mn和氧空位的存在产生的能量禁带。此外,将Mn掺杂进TiO2晶格后,Mn-TiO2催化剂的颜色加深。同样,磷酸改性并未改变催化剂本身的UV-Vis吸收性能,因此Mn-TiO2和P-Mn-TiO2的吸光性能仅因Mn的掺杂得到了明显的增强。值得注意的是,由于Mn掺杂后,带宽减小,光生载流子更容易复合,降低光催化性能。
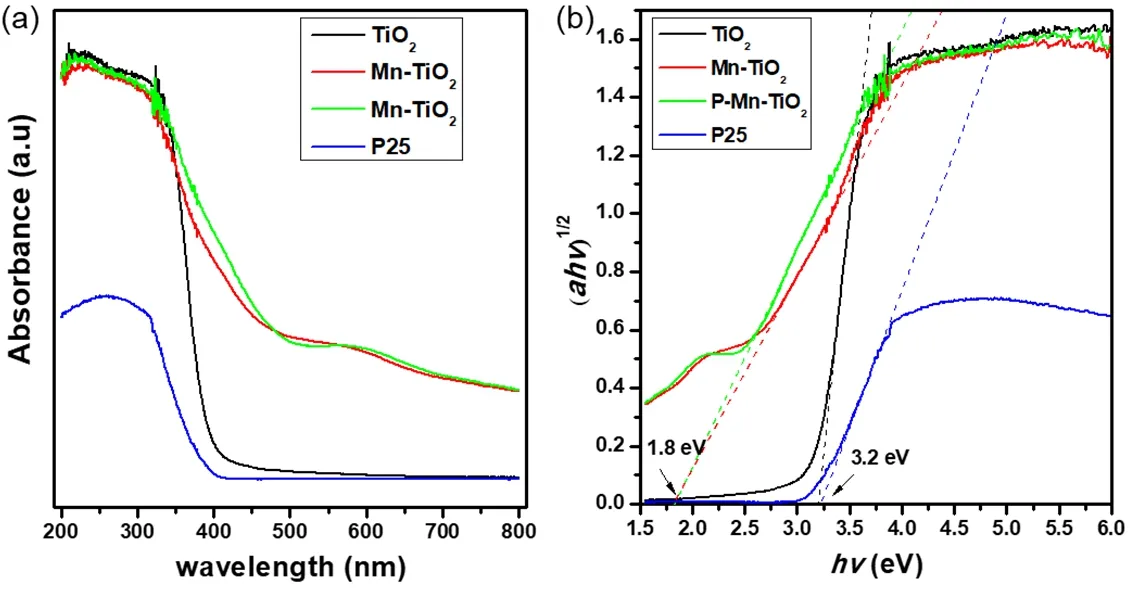
图6 不同催化剂的UV-Vis图谱和(αhv)1/2 vs. hv曲线Fig. 6 UV-Vis diffuse reflectance spectra and plots of (αhv)1/2 vs. photo energy of different samples.
3.2 VUV-PCO甲苯降解性能
图7所示为不同催化剂在VUV光照下降解甲苯的性能。其相应的O3和COx产生浓度分别列于图8和图9。
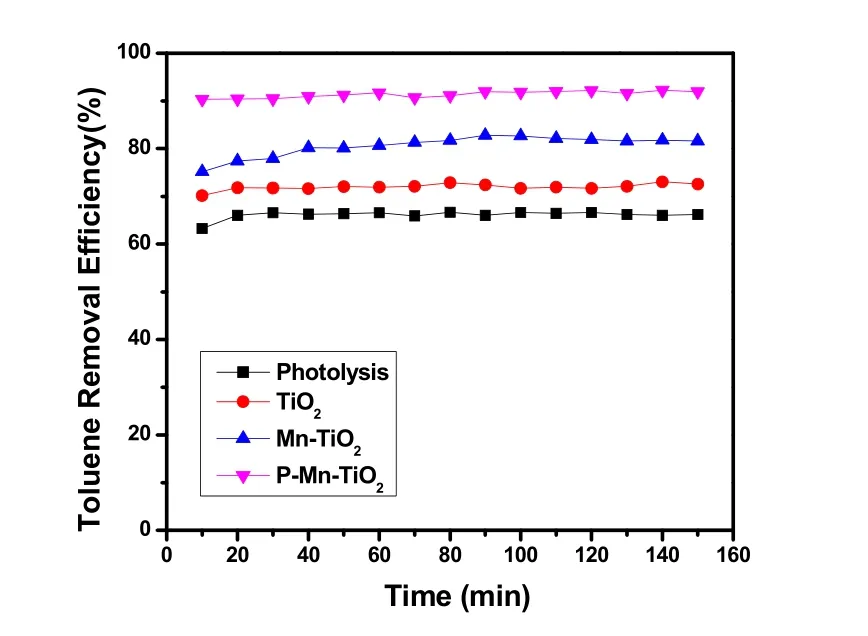
图7 不同催化剂在VUV光照下甲苯的去除效率Fig. 7 Toluene removal efficiency over different samples under VUV irradiation.
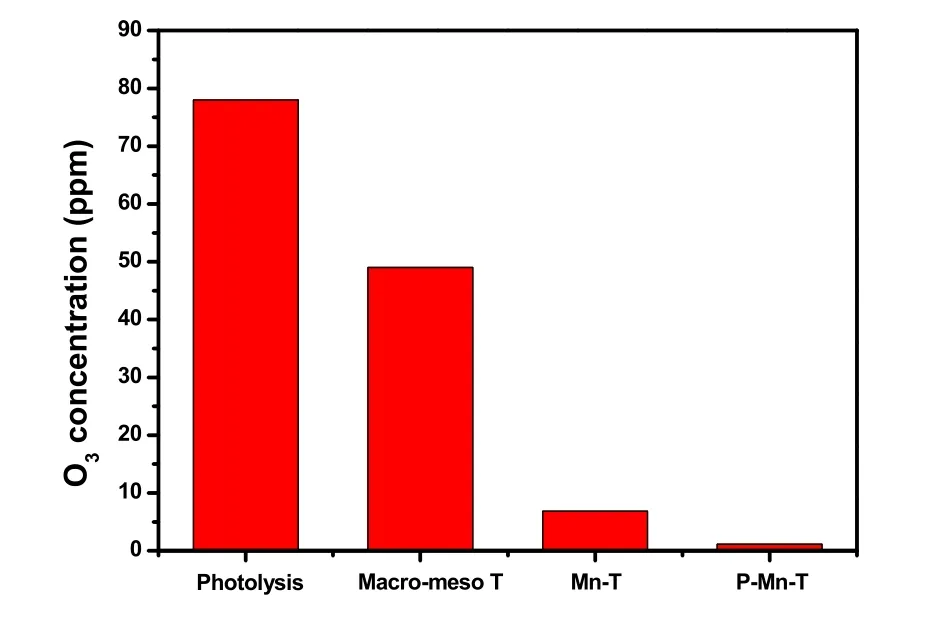
图8 VUV-PCO体系中不同催化剂产生的臭氧浓度Fig. 8 Outlet O3 concentration of different samples in the VUV-PCO system.

图9 VUV-PCO体系中不同催化剂产生的COx浓度Fig. 9 COx concentration of different samples in the VUV-PCO system.
如图7,不同催化剂的甲苯去除效率顺序依次为:VUV photolysis < VUV + TiO2< VUV + Mn-TiO2< VUV + P-Mn-TiO2。在甲苯初始浓度为40 ppm时,单独VUV光解甲苯的去除率稳定在65%左右,这主要来源于VUV紫外光内185 nm所含有的高能光子光解甲苯和激发氧气和水产生活性氧物种及羟基氧化所致。原始未负载活性组分的介孔TiO2在VUV光照150 min后,其对甲苯的去除率保持并稳定在70%左右,略高于光解,说明催化剂本身催化性能不显著,这可能是由于本实验是在动态气相反应,其气体流速快,催化剂接触时间短,因此整体的污染物降解效率要低于静态气相反应。Mn-TiO2在光照150 min后,其甲苯去除率约为80%,高于VUV光解和TiO2,说明Mn掺杂后,其催化氧化降解甲苯活性提高。在我们前期的研究中发现,VUV光解后残余未反应的甲苯在Mn-TiO2作用下,经历了臭氧催化氧化降解。当在Mn-TiO2催化剂表面进行磷酸修饰后,其催化氧化性能有显著提高。如图所示,P-Mn-TiO2在VUV光照150 min后,其甲苯去除率能稳定在90%左右。研究表明36,磷酸表面修饰,可替换掉表面部分羟基基团,同时增强催化剂表面对O2、O3等氧物种的吸附,加速光生电子的与氧物种反应,提高光生载流子分离效率,促进光生空穴生成未束缚的流动态羟基自由基,或直接参与污染物的氧化,高效地促进气态甲苯的降解。
但是VUV单独光解空气时会产生大量臭氧:
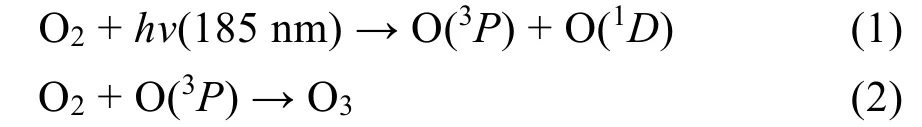
如图8所示,VUV单独光解甲苯时,体系中约有80 ppm的臭氧产生。而臭氧作为良好的电子受体,可以有效的捕获电子,抑制光催化反应中光生电子和空穴的复合,并延长空穴的寿命39,40。但是TiO2存在下,体系对臭氧的消除不彻底,仍然有50 ppm左右的臭氧残留,释放出来。而大量研究发现41,锰氧化物对臭氧的分解和利用率高,能产生大量氧空位,吸附催化臭氧生成活性氧物种,降解气态污染物。因此,Mn掺杂的光催化剂Mn-TiO2同样对臭氧有高效的分解和利用能力,体系中臭氧的出口大大减少到约6 ppm。在XPS分析中我们检测到Mn3+的存在,并认为Mn3+/Mn4+离子掺杂进TiO2的晶格有助于生成氧空位,而臭氧催化分解及氧化性能又和催化剂表面的氧空位循环有关42。首先,臭氧会通过将一个氧原子插入到氧空位的方式吸附到催化剂表面,此时,臭氧上会有电子传递到氧空位上连接的氧原子,产生一个∙O−和O2(公式(3))。随后,产生的∙O−和另外一个臭氧分子反应,生成一个O2和过氧∙O2−(公式(4))。生成的∙O2−活性氧物种一方面可以直接氧化甲苯或者和H2O反应生成∙OH(公式(6)),另一方面又可以分解释放一个氧空位和氧气(公式(5)),参与到下一轮臭氧的分解循环中。在图8中,介孔二氧化钛使臭氧出口浓度从80 ppm降为50 ppm,再次证明二氧化钛能有效分解臭氧,但是在二氧化钛中掺杂少量的Mn就能使臭氧的出口浓度一直保持6 ppm,同时磷酸修饰后,臭氧出口浓度保持约为1-2 ppm左右,再次证明磷酸修饰后,催化剂表面更易吸附氧气、臭氧等,而光生电子与吸附的臭氧反应(公式(7、8)),从而引入了更多的活性表面氧物种,进一步提高甲苯氧化性能,消除体系臭氧量(公式(9))。

在VUV-PCO体系中,甲苯的降解主要来源于VUV直接光解,光催化氧化和臭氧催化氧化联合作用。一般认为,在臭氧催化氧化反应中,活性氧物种(O−/O2−)的生成是其催化效率的决速步骤43,同时,多孔材料TiO2的介孔和大比表面积又能为活性物种和臭氧提供吸附和反应位点。因此,在PMn-TiO2作用下,光催化和臭氧催化氧化协同作用能够有效提高甲苯的降解效率。
图9所示为甲苯在不同催化剂作用下氧化生成的COx浓度。从图中可以看出,P-Mn-TiO2催化剂相对于其他材料不仅有最高的臭氧去除率,其甲苯降解率及碳氧化物生成量都是最高。不同催化剂的碳氧化物出口浓度顺序依次为:VUV photolysis < P25 < TiO2< Mn-TiO2< P-Mn-TiO2,和甲苯的降解效率一致。值得注意的是,各个催化材料的CO出口浓度都接近于25 ppm,与光解产生的CO浓度相差不大,说明体系中CO主要来源于VUV光解。
3.3 催化剂调控
为了优化催化剂的性能,提高催化剂表面的活性位数目,我们对复合催化剂P-Mn-TiO2的组分进行了调控,并相应测试了其甲苯降解性能。如图10所示,醋酸锰前驱体浓度为0.05 mol·L−1时,Mn-TiO2的甲苯去除率相对最高,为80%左右,其次是浓度为0.1 mol·L−1醋酸锰所合成的催化剂,甲苯去除率为75%左右,说明在一定范围内,Mn负载量的增加有利于提高催化剂降解甲苯的性能,但Mn负载量过高反而导致甲苯去除率的下降。同样,在图10b中,可以发现,磷酸前驱体浓度为0.05 mol·L−1时,其P-Mn-TiO2的甲苯去除性能最优,高于90%。而在其他磷酸浓度下处理的催化剂,其甲苯降解性能相差不多,约为85%左右,但仍然高于Mn-TiO2,进一步说明磷酸根修饰有助于提高催化剂的甲苯降解性能。根据以上分析,在催化剂上掺杂和修饰合适浓度的Mn和[PO4]3−阴离子基团有助于提高催化剂的活性位点,增强对氧气、臭氧等的吸附和转化性能,从而提高其催化氧化甲苯性能。
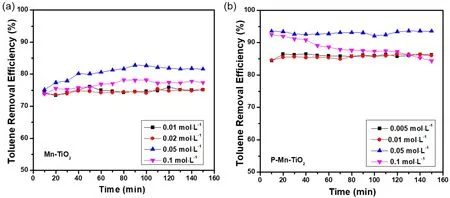
图10 (a)不同Mn含量和(b)磷酸浓度下催化剂在VUV光照下甲苯的去除效率Fig. 10 Toluene removal efficiency over samples at (a) different Mn loading and (b) different phosphate loading under VUV irradiation.
4 结论
在本工作中我们成功合成了多功能P-Mn-TiO2催化材料,将其应用于VUV-PCO体系中,光催化氧化甲苯的同时消除并利用臭氧。相对于传统UV体系,VUV真空紫外灯的甲苯降解性能极具优越性。磷酸修饰和Mn掺杂的介孔二氧化钛(PMn-TiO2)具有高效甲苯去除效率(90%)。在P-Mn-TiO2催化材料的作用下,体系中的臭氧副产物几乎能被完全消除,同时转化为活性氧物种氧化甲苯。分析发现,Mn-O-Ti化学键间强烈的相互作用有助于降低氧空位的生成势能,同时,Mn掺杂进Ti的晶格能够提高TiO2的吸光性能,UV-Vis中出现红移现象。因此催化剂Mn3+的浓度和含量以及从体相传递到表相的氧空位含量显著增加,更多的臭氧等氧物种吸附到氧空位上反应,生成活性氧物种∙O2−、羟基自由基∙OH等。此外,P-Mn-TiO2光催化材料表面的磷酸功能基团,同样能高效吸附氧气并转化臭氧,提高催化剂光生电子分离效率,并生成大量活性氧物种。因此,在本VUV-PCO体系中,存在光解、光催化和臭氧催化氧化之间的协同作用,首先真空紫外灯含有能力约为8%的185 nm紫外光,不仅能直接光解气态污染物,还能激活空气中的水蒸气和氧气,产生∙OH、∙O和O3等活性氧物种间接氧化气态污染物。同时VUV真空紫外灯发射的185和254 nm紫外光均可被光催化剂吸收,在催化剂TiO2表面发生光催化反应,产生光生电子和空穴,进一步转变为强氧化性物种,用于进一步氧化甲苯。同时,由于我们经多孔TiO2表面进行Mn和磷酸修饰,使催化剂表面有大量氧空位和活性氧物种,有效吸附氧气和臭氧,将其转化为活性氧物种,氧化甲苯的同时,可以消除副产物臭氧的排放。
猜你喜欢
杂志排行

物理化学学报的其它文章
- 基于密度泛函理论下H2S 在单原子催化剂V/Ti2CO2 上的分解机理研究
- 富含缺陷的2D/2D 异质结促进光催化清洁能源转化
- Development of Iron-Based Heterogeneous Cocatalysts for Photoelectrochemical Water Oxidation
- 二维光催化材料电子结构和性能调控策略研究进展
- Defect Engineering in Two-Dimensional Graphitic Carbon Nitride and Application to Photocatalytic Air Purification
- Non-Noble-Metallic Cocatalyst Ni2P Nanoparticles Modified Graphite-Like Carbonitride with Enhanced Photocatalytic Hydrogen Evolution under Visible Light Irradiation